Syndromic congenital heart defects (CHDs) that are associated with multisystem congenital anomalies comprise 20–30% of the total CHD population.1) Advances in molecular diagnostics have deepened our understanding of underlying genetic etiologies in syndromic CHDs, and have contributed to the expanding list of differential diagnoses. However, recognizing rare and phenotypically variable forms of syndromic CHD remains a challenge.
Copy number variations (CNVs) are the cause of 10–20% of syndromic CHDs. Chromosomal microarray (CMA) testing, which aims to detect CNVs in a genome-wide manner, has thus recently earned a role as the first-line diagnostic tool in syndromic CHD diagnosis.2) CMA aids our way through complicated differential diagnoses in an unexplained syndromic CHD circumstance.3)
We here report on two patients successfully diagnosed by CMA testing to have Mowat–Wilson syndrome (MWS). MWS is one of the very rare syndromic CHDs, the diagnosis of which may often be delayed, based solely on clinical findings. We also discuss the approach towards syndromic CHDs in the era of molecular diagnostics.
Patient 1
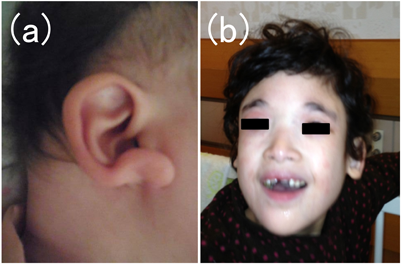
The patient is a 3-year-old girl born as the second child of consanguineous parents. Her CHD diagnosis of pulmonary atresia with ventricular septal defect, and right aortic arch was made antenatally. Delivery was uneventful and she was admitted to the neonatal intensive care unit (NICU). Due to early feeding difficulties, she required a feeding tube and remained tube-dependent until one year of age. After undergoing an initial Blalock-Taussig shunt palliation, she underwent repair of her CHD during infancy. She had a characteristic facial appearance with medially flared eyebrows, hypertelorism, telecanthus, and uplifted ear lobes (Fig. 1 (a)). Conventional karyotyping revealed inv(9)(p12q13), which was not diagnostic for her manifestations. She also tested negative for 22q11.2 deletion. Given her ear anomalies and iris colobomas along with her CHD diagnosis, CHARGE syndrome was considered as her primary diagnosis.
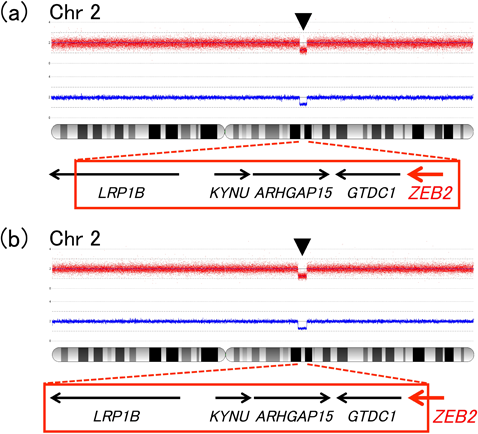
At age 3, she could walk a couple of steps with aid, and use very few, but intelligible words. By then, microcephaly had become a more prominent feature, and the facial gestalt prompted us to reconsider her diagnosis. Since she lacked some classical features of MWS, such as Hirschsprung disease, corpus callosum defects and urogenital anomalies, instead of performing MWS-targeted ZEB2 gene analyses, we selected CMA testing as our initial approach. CMA revealed a ZEB2-containing 3.7 Mb deletion (arr[hg19] 2q22.1q22.3 (141,783,763–145,531,555)×1), confirming the diagnosis of MWS (Fig. 2(a)). Parental testing determined that the deletion was de novo in origin.
Patient 2
The patient is an 11-year-old girl who was born as the third child of consanguineous parents. A heart murmur and stridor was heard after birth and the patient was transferred to the NICU, where the diagnosis of pulmonary artery sling, secundum atrial septal defect and tracheal stenosis was made. Her CHD was repaired during her initial hospitalization. She had a characteristic facial appearance with medially flared eyebrows, hypertelorism, telecanthus, uplifted ear lobes and a pointed chin (Fig. 1(b)). At age 3, a prolonged seizure episode occurred, accompanied by an abnormal electroencephalogram pattern of diffuse, high amplitude, spike-and-wave activities. Together with severely impaired speech, frequent laughing, happy disposition and facial features, her manifestations were suggestive of Angelman syndrome. However, genetic workup, including copy number analysis of the 15q11–q13 region, methylation analysis of SNRPN and UBE3A sequencing had all turned out negative, obscuring a definite diagnosis.
Later in life, microcephaly and severe developmental abnormalities became gradually manifest. At age 11, she still could not stand unaided and no intelligible words could be heard. In search of an etiology explaining her multiple anomalies, CMA testing was performed. CMA testing established a molecular diagnosis of MWS by revealing a 4.5 Mb deletion (arr[hg19] 2q22.1q22.3 (140,569,236–145,155,731)×1), involving exon 9–10 of the ZEB2 gene (Fig. 2(b)). Parental testing confirmed de novo occurrence of the deletion.
We report on two patients with CHD and concomitant unexplained multisystem anomalies suggestive of an underlying genetic disorder. The genetic etiology of MWS was successfully diagnosed by CMA testing. MWS is a recently established, thus under-recognized, congenital anomaly syndrome, the awareness of which needs to be raised among pediatric cardiologists.
Patients with MWS present with a characteristic facial appearance, including medially flared eyebrows, hypertelorism, telecanthus, a broad nasal bridge, a pointed chin, and uplifted ear lobes. Intellectual disability, especially severely impaired speech, microcephaly, epilepsy, agenesis of the corpus callosum, Hirschsprung disease, urogenital anomalies, and congenital heart disease are the classic manifestations of MWS.4) However, MWS diagnosis is at times challenging; the rarity, phenotypic variability and phenotypic overlap with other congenital disorders may hinder clinical recognition of the syndrome.
Among the highly variable spectrum of CHDs seen in MWS, pulmonary artery sling, an extremely rare CHD itself, seems a particular association, recurring in numerous MWS cases reported in the literature.5, 6) Previous studies have implicated the importance of ZEB2, the MWS-causing gene in the formation of neural crest derivatives. The spectrum of cardiovascular anomalies affecting patients with MWS are likewise thought to be the consequence of a cardiac neural crest induction, migration, and/or differentiation defect. While the cardiovascular malformations seen in ZEB2-ablated mouse models do not fully recapitulate those observed in human patients,7) by way of the quail-chick chimera technique and retroviral LacZ reporter introduction, studies on chick embryos have elegantly revealed the contribution of neural crest cells to the developing proximal pulmonary arteries.8) Thus, pulmonary valve and pulmonary artery abnormalities, as found in our two patients, may well be clues to phenotypic recognition of MWS.5) Ocular anomalies, such as colobomas seen in Patient 1, have been observed in other patients with MWS,9) and may also facilitate diagnosis in the absence of some classic features of MWS.
The genetic cause of MWS is haploinsufficiency of the ZEB2 gene at 2q22. While the majority of disease-causative defects are nonsense or frameshift nucleotide mutations, around 20% are more gross deletions within or including the ZEB2 gene,10, 11) which could potentially be overlooked by traditional sequencing technologies. Since our patients lacked some of the classical features of MWS, such as Hirschsprung disease and corpus callosum defects, we were not capable of narrowing down the differential diagnosis, and thus withheld performing ZEB2-targeted sequencing or dosage analysis. Furthermore, the clearly syndromic nature of their CHD phenotype prompted us to perform CMA testing as an initial approach, in search of CNVs responsible for the patients’ conditions. Our approach was contrary to the genetic testing strategy proposed by Saunders et al.,12) but as we have shown, workup may be individualized according to the patients’ presentation and strength of clinical suspicion.
For experienced dysmorphologists, establishing the diagnoses of syndromic CHDs including MWS may be quite straightforward. However, in Japan, there is a shortage of genetics and dysmorphology experts, and in such a situation clinical recognition of rare genetic syndromes is still a challenge. This background may lead us to rely more heavily on increasingly available genetic diagnostic tools. In the meantime, the cost and time consumed in sequencing genomes will steadily become reduced. Furthermore, the diagnostic yield of genetic testing may even surpass that of a thorough exam by a well-trained dysmorphologist.3) It is easy to imagine that in the near future, even more genome-wide genetic testing will be performed, and accordingly more genetic diagnoses will be made.
On the other hand, there are drawbacks to taking high-resolution, genome-wide genetic testing approaches. Experts have raised caution on interpreting CMA testing results, especially about overestimating the causality of de novo CNVs.13) Along with well-defined, recurrent, disease associated mutations, CMA testing usually reveals many unexpected findings, and variants of unknown significance. Confirming the causality of each individual variation is challenging. We clinicians rely on recurrence of identical variants in patient cohorts, and consistency with in vitro functional studies, in establishing a causality relationship of a novel CNV. Being de novo of origin, or involving genes known to function during embryological development, raises the possibility of the CNV being pathogenic, however, it is not a golden rule.
Pediatric cardiologists must be aware of the strengths and shortcomings of each available genetic testing modality and be well prepared for the era of molecular diagnostics. For the sake of our patients and their families, improving genetics literacy among pediatric cardiologists is an urgent issue.