Troponin T2 (TNNT2) gene mutations are known as one of the causes of familial hypertrophic cardiomyopathy. We present a 1-year-old Japanese girl who presented with sudden death and was found to have low activity of the cardiac mitochondrial respiratory transport chain complex (MRTCC) I, which can cause acute cardiac dysfunction. The patient had no causative gene mutations suggestive of a mitochondrial disorder, but was found to have a Phe110Ile missense mutation (c.358T>A; p.F110I) in the cardiac TNNT2 gene.
A 1-year-old Japanese girl with a three-hour history of diaphoresis and emesis, was brought to our emergency room (ER) by her mother. She was diagnosed with acute gastroenteritis (Fig. 1) and was discharged back to her home. Five hours later, the patient had recurrent emesis, which progressed to apnea. The child was driven by her mother back to the same ER. Upon arrival, the child was in cardiopulmonary arrest. Her initial electrocardiogram finding revealed asystole. Unfortunately, despite an hour of cardiopulmonary resuscitation, the patient died.
The patient had suffered with hand-foot-and-mouth disease two weeks prior to her demise. She had otherwise been well and had achieved her normal developmental milestones. She had no other significant illnesses during her life and was taking no medications. She had no allergies.
Her oldest brother was born with a cleft palate and syndactyly and suffered with atopic dermatitis. Her second oldest brother suffered with asthma. Moreover, a maternal cousin died as a neonate secondary to renal failure.
A blood gas analysis on arrival was significant for a mixed acidosis. The laboratory data demonstrated liver enzyme elevations (see Table 1). For investigations into the cause of death, postmortem examinations were undertaken. Coxsackievirus A16 and Human herpesvirus 6B were detected in her pharynx, whereas Adenovirus Type 2 and Coxsackievirus A16 were detected in her stool, respectively. These viruses were not detected in her serum, pleural effusions, pericardial fluid, lungs or liver. Respiratory syncytial virus and Human metapneumovirus were both negative on rapid antigen testing of her nasal cavity. The following organisms were detected in various locations: Staphylococcus epidermidis in her blood, Enterobacter cloacae complex in her pharynx, Haemophilus influenza in her nasal cavity, and Candida albicans in her endotracheal tube.
Table 1 The laboratory data on arrival at our hospital| Blood gas analysis |
|---|
| pH | 6.499 (7.37–7.45) |
| PaCO2 | 93.2 mmHg (33.0–45.0 mmHg) |
| PaO2 | 24.9 mmHg (70–115 mmHg) |
| HCO3− | 7.2 mmol/L (22.0–28.0 mmol/L) |
| Base Excess | −30.6 mmol/L (−2.0–2.0 mmol/L) |
| Na+ | 145 mEq/L (137–147 mEq/L) |
| K+ | 7.8 mEq/L (3.5–5.0 mEq/L) |
| Cl− | 107 mEq/L (97–108 mEq/L) |
| Ca2+ | 2.63 mEq/L (2.30–2.58 mEq/L) |
| Blood glucose | 210 mg/dL (70–109 mg/dL) |
| Lactate | 204 mg/dL (3.7–16.3 mg/dL) |
| Complete blood count |
|---|
| WBC | 11,200/µL (4,000–9,000/µL) |
| Hb | 11.0 g/dL (11.5–15.0 g/dL) |
| Platelets | 3.16×105/µL (1.4×105–3.5×105/µL) |
| Biochemistry |
|---|
| AST | 116 U/L (13–33 U/L) |
| ALT | 58 U/L (6–27 U/L) |
| LDH | 830 U/L (119–229 U/L) |
| CK | 1,759 U/L (45–163 U/L) |
| CRP | Unmeasurable because of lack of sample amount |
| ALT, alanine transaminase; AST, aspartate transaminase: Ca2+, Calcium; CK, creatinine kinase; Cl−, Chloride; CRP, C-reactive protein; Hb, hemoglobin; HCO3−, bicarbonate; LDH, lactate dehydrogenase; K+, Potassium; Na+, Sodium; WBC, white blood cells. |
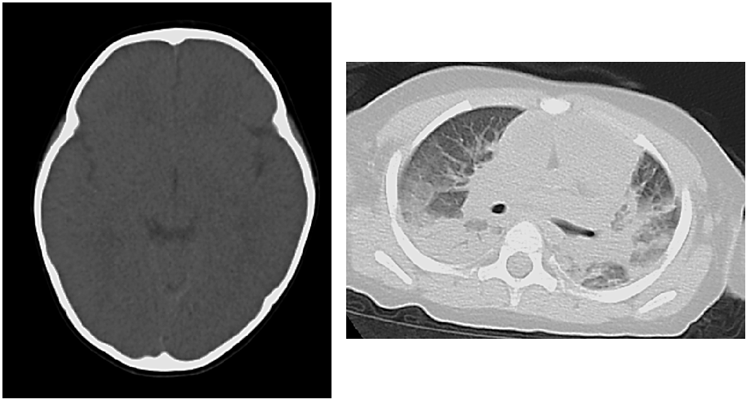
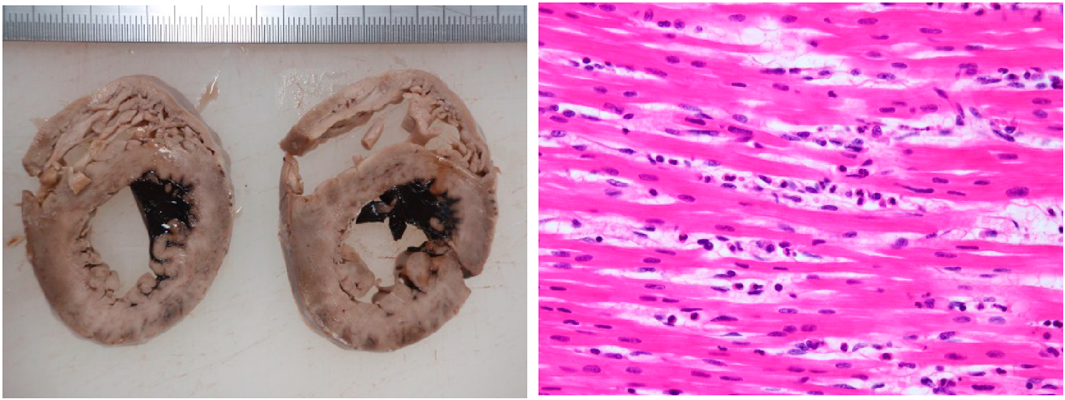
A blood acylcarnitine analysis was normal. This ruled out an organic acid or fatty acid metabolic abnormality. Computed tomography findings revealed cerebral edema, pulmonary edema and atelectasis, which indicated postmortem changes (Fig. 2). Nine hours after death, a pathologic examination revealed pulmonary edema, alveolar hemorrhage, and non-suppurative pericarditis with lymphocyte invasion into the epicardium. There was mild neutrophil infiltration into the myocardium, but no findings of myocardial necrosis or edema. There were no pathological findings of virus-induced changes, fulminant myocarditis, myocyte disarray, or myocardial hypertrophy (Fig. 3). We did not perform an electron microscopic study.
For investigations of the cause of acute cardiac dysfunction, we measured the activity of the MRTCC (see Table 2). The activity of the cardiac MRTCC I was reduced. As a result, we analyzed target resequencing by use of a genetic testing panel for mitochondrial diseases. The child had no causative gene mutations revealing a mitochondrial disorder. However, the child had a Phe110Ile missense heterozygous mutation in the cardiac TNNT2 gene (c.358T>A; p.F110I (NM_000364.2; NP_000355.2)). A TNNT2 mutation is one of the causes of familial hypertrophic cardiomyopathy (FHCM). We analyzed the patient’s family genotype, and identified her father and second oldest brother to have the identical gene mutation. Their electrocardiograms and 2-dimension echocardiograms were normal. They had experienced no episodes of acute cardiac dysfunction or symptoms with regard to a mitochondrial disorder in the past.
Table 2 Activity of the mitochondrial respiratory chain
complex I–IV| Heart (%) | Co I | Co II | Co II+III | Co III | Co IV | CS |
|---|
| Control* | | | | | | |
|---|
| % of normal | 39.8 | 59.4 | 39.6 | 31.7 | 30.4 | 60.1 |
| CS ratio (%) | 62.2 | 98.9 | 57.5 | 52.4 | 48.5 | |
| Co II ratio (%) | 62.3 | | 56.9 | 53.3 | 48.7 | |
| Patient | | | | | | |
|---|
| % of normal | 4.1 | 44.8 | 64.8 | 50.5 | 50.4 | 59.5 |
| CS ratio (%) | 6.4 | 75.3 | 95.0 | 84.2 | 81.1 | |
| Co II ratio (%) | 8.4 | | 123.4 | 112.4 | 107.0 | |
| Liver (%) | Co I | Co II | Co III | Co IV | CS | |
|---|
| Control* | | | | | | |
|---|
| % of normal | 58.9 | 77.8 | 93.5 | 83.1 | 93.7 | |
| CS ratio (%) | 60.2 | 79.8 | 95.1 | 84.6 | | |
| Co II ratio (%) | 73.8 | | 117.7 | 84.5 | | |
| Patient | | | | | | |
|---|
| % of normal | 27.6 | 87.8 | 47.6 | 132.0 | 110.3 | |
| CS ratio (%) | 23.9 | 76.4 | 41.1 | 114.1 | | |
| Co II ratio (%) | 30.7 | | 53.2 | 119.1 | | |
| *Control samples were derived from the living tissue of liver transplant donors (n=12) and the hearts of patients with congenital heart disease in Japanese and Australian children aged between 0–10 years old (n=7). The proportion of males to females was 1 : 1. Co I, complex I; Co II, complex II; Co III, complex III; Co IV, complex IV; CS, citrate synthase. |
A TNNT2 mutation has an autosomal dominant inheritance, is one of the causes of FHCM, dilated cardiomyopathy (DCM), restrictive cardiomyopathy (RCM), and left ventricular non-compaction (LVNC). The chromosomal location of the mutation is at 1q32.1.1) The Phe110Ile mutation has been reported to have comparatively slight hypertrophy or apical hypertrophy.2) Despite the lack of myocyte hypertrophy, disarray, or myocardial fibrosis, the Phe110Ile mutation may cause impaired acute exercise tolerance. An increased Ca2+ sensitivity of force has been observed in F110I-TnT-reconstituted human cardiac muscle preparations. Transgenic (Tg) mice expressing F110I-TnT’s ratios of ATPase/force (energy cost) at various Ca2+ concentrations were dramatically higher compared with non-Tg fibers. This increase in energy cost most likely results from a decrease in force per myosin cross-bridge.3)
MRTCC is a five-protein complex within the mitochondrial inner membrane. MRTCC I–IV are the electron transport chain, whereas V is adenosine triphosphate synthase. They play an important role in cellular respiration. Congenital hypofunction of MRTCC is a cause of mitochondrial disease. Mitochondria exist in all cells except erythrocytes and as a result, mitochondrial disease can present with various symptoms. However, there is a specific disease type which can present with single organ dysfunction such as cardiomyopathy. Recently, many causative mutations have been found in the MRTCC subunits and MRTCC forming factors, respectively.4)
There have been no previous reports that indicate TNNT2 mutations result in low activity of MRTCC I. However, there was a report of a TNNI3 mutation and MRTCC I. This was a heterozygous mutation c.575G>A (p.R192H) in TNNI3 (NM_000363), which was previously identified in patients with restrictive cardiomyopathy. Electron microscopic examination also revealed abnormally shaped mitochondria with concentric cristae. Because these genes and loci are not directly linked to the respiratory chain complex, they considered the mitochondrial respiratory chain complex deficiencies are secondary.5) In an animal model there were increased numbers of mitochondria with evidence of mitochondrial degeneration as evidenced by small mitochondria with loss of well-defined membranes and cristae.6)
Pathologic examination of our patient indicated no findings of FHCM, DCM, RCM, or LVNC, respectively. Clinically, we speculate that the child’s diaphoresis and emesis were symptoms of low cardiac output from acute cardiac dysfunction. Both TNNT2 mutations and MRTCC, might have resulted in sudden death in this patient. The cause of acute cardiac failure leading to cardiac arrest can be potentially explained by the intolerance for increased cardiac stress following a viral infection, which resulted from higher energy cost (the ratios of ATPase/force) secondary to the Phe110Ile mutation, and by a lack of myocardial energy which resulted from decreased activity of MRTCC I. However, it is unclear if her death was due to the mutation.
Finally, her father and second older brother exhibited the same mutation, but they were asymptomatic with regard to HCM. However, FCHM has an age-related penetrance, so we have recommended annual cardiac examinations for the two affected family members.
Our case is the first report of a patient with a TNNT2 mutation and low activity of cardiac MRTCC I. It is unclear that the child’s mutation resulted in her death.
謝辞Acknowledgments
We would like to thank Dr. Joel Branch, Shonan Kamakura General Hospital, Japan, for his kind English language review and correction of this case report. In addition, we thank Kei Murayama, Department of Metabolism, Chiba Children’s Hospital, Chiba Prefecture, Japan, for measuring the activity of MRTCC and for performing genetic testing.
Conflicts of Interest
The authors declare that there is no conflict of interest.
引用文献References
1) Online Mendelian Inheritance on Man: An Online Catalog of Human Genes and Genetic Disorders. OMIM #191045, TROPONIN T2. https://www.omim.org/entry/191045 (as of January 16, 2019)
2) Anan R, Shono H, Kisanuki A, et al: Patients with familial hypertrophic cardiomyopathy caused by a Phe110Ile missense mutation in the cardiac Troponin T gene have variable cardiac morphologies and a favorable prognosis. Circulation 1998; 98: 391–397
3) Olga H, Danuta S, Bjorn K, et al: F110I and R278C Troponin T mutations that cause familial hypertrophic cardiomyopathy affect muscle contraction in transgenic mice and reconstituted human cardiac fibers. J Biol Chem 2005; 280: 37183–37194
4) Mimaki M: Diversity of mitochondrial disorders: What child neurologists should know. No To Hattatsu 2018; 50: 7–16 (in Japanese)
5) Kohda M, Tokuzawa Y, Kishita Y, et al: A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet 2016; 12: e1005679
6) Jil T, Timothy H, Bradley P, et al: Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J Clin Invest 1998; 104: 469–481