Propionic acidemia is an autosomal recessive hereditary disease that caused by a defect in propionyl-CoA carboxylase (PCC) present in the mitochondria. PCC is composed of an alpha subunit and a beta subunit, which are encoded by the PCCA and PCCB genes, respectively. Propionyl-CoA, a substrate for PCC, is derived from the metabolism of amino acids (e.g., isoleucine, valine, threonine, and methionine), odd-chain fatty acids, cholesterol side chains, and intestinal flora. Propionyl-CoA is converted to methylmalonyl-CoA by PCC, changed to succinyl-CoA, and enters into the citric acid cycle.1) Thus, in patients with PCC deficiency, organic acids such as propionic acid accumulate from the start of feeding in the neonatal period, leading to the development of metabolic crisis. There is also a delayed type that does not result in metabolic crisis in the neonatal period. This type is diagnosed based on symptoms, such as mental retardation or recurrent vomiting. Cardiomyopathy is present in 9–23% of patients with propionic acidemia;1) however, this condition is often overlooked especially in the delayed type of DCM. In this article, we report a case of dilated cardiomyopathy (DCM) in a patient with mental retardation and epilepsy, that was diagnosed as propionic acidemia through diagnostic work-up of cardiomyopathy.
The case was a 14-year-old female who was born via Cesarean section with a 40-week gestational age and birth weight of 2,902 g, as the second child of non-consanguineous parents. Her mother was diagnosed with gestational diabetes during pregnancy of the patient. Her father, and the older and younger brothers were healthy. Her neonatal period was uneventful. At the age of 2 years, she experienced a febrile seizure. Developmental delay and mild mental retardation were identified during attendance at school. At the age of 12 years, she experienced an afebrile seizure. Electron encephalography showed polyspike-and-slow wave complex in both central and temporal regions; hence, she was diagnosed with epilepsy. She experienced menarche when she was 13 years old. She had another episode of an afebrile seizure in the same year; however, the parents did not provide consent to treat her epilepsy. At this point, her resting heart rate was normal. At the age of 14 years, tachycardia (129 beats per minute) was noted during a cardiac screening test conducted in school and she visited our hospital. She was asymptomatic and obese. Her body weight was 67.6 kg and her height 156.7 cm (body mass index 27.5). Her heart sound was regular without significant murmur. There was no enlargement of the liver or edema in the lower legs, and her extremities were not cold.
Laboratory tests showed mild anemia with hemoglobin 10.5 g/dL, hematocrit of 35.9%, and high level of brain natriuretic peptide (62.9 pg/mL). Her liver and renal function were normal (aspartate aminotransferase 12 U/L, alanine aminotransferase 8 U/L, lactate dehydrogenase 168 U/L, urea nitrogen 10 mg/dL, creatinine 0.49 mg/dL). The levels of lactate and ammonia were within the normal range (lactate 17.6 mg/dL, ammonia 45 µg/dL). Her lipid profile was also normal (total cholesterol 173 mg/dL, triglyceride 73 mg/dL). The fasting blood glucose level was slightly high (117 mg/dL), although hemoglobin A1c was within the normal range (5.2%). There was no metabolic acidosis with venous blood gas at pH 7.428, PCO2 36.7 mmHg, HCO3− 23.8 mmol/L.
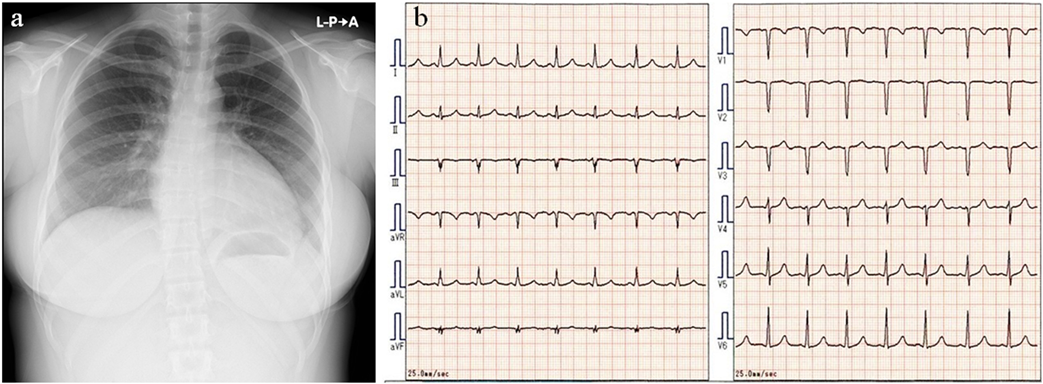
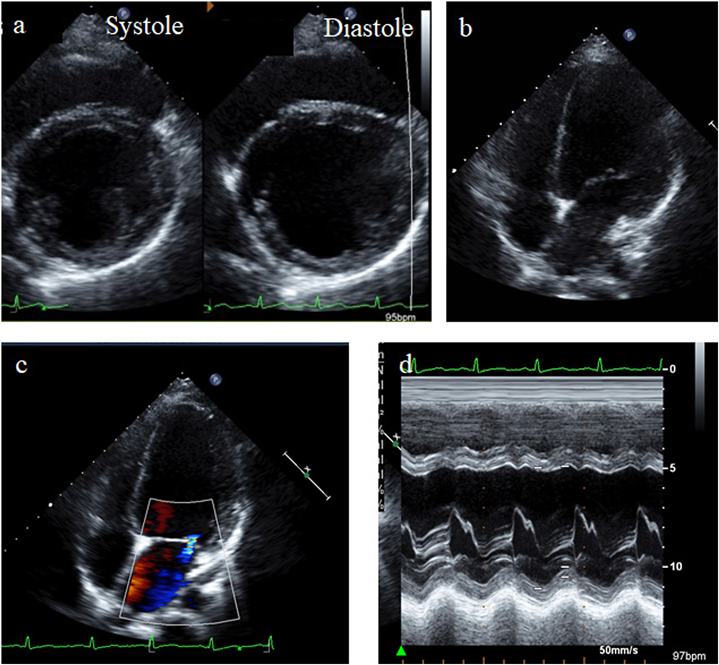
Chest X-ray showed cardiac enlargement with the cardiothoracic ratio of 53% (Fig. 1a). Electrocardiogram showed poor R wave progression and the corrected QT interval was 436 ms (Fig.1b). Echocardiography showed dilated left ventricular size (Fig. 2a, b) with an end diastolic diameter of 65.0 mm (Z-score +3.58), moderate mitral valve regurgitation (Fig.2c), and left ventricular ejection fraction of 35% (Fig. 2d). These findings were compatible with DCM.
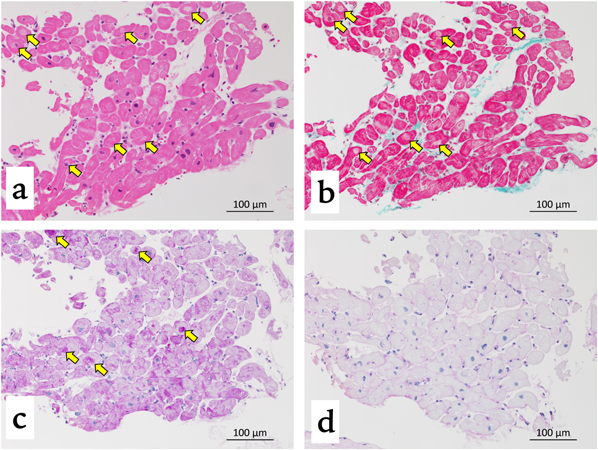
Endomyocardial biopsy showed hypertrophy of cardiomyocytes and vacuoles, which were located around the nuclei of some cardiomyocytes under light microscopy. The vacuoles were positive for periodic acid-Schiff (PAS) staining and negative for PAS-diastase staining (Fig. 3). Inflammatory cell infiltration or fibrosis was not observed. Electron microscopy showed an increase in the number of mitochondria within myofibrils, and the presence of large mitochondria with swirling cristae (Fig. 4). Moreover, there were lipid droplets and glycogen vesicles on the cardiomyocytes under electron microscopy.
The acylcarnitine analysis of dried blood spot showed an increase in propionylcarnitine (12.3 µmol/L, cut-off value 4.0 µmol/L) and the propionylcarnitine/acetylcarnitine ratio (0.39, cut-off value 0.25). The level of free carnitine was normal (47.5 µmol/L, normal range 36.0–74.0 µmol/L). Urine organic acid analysis showed an increase in methylcitric acid and 3-hydroxypropionic acid, without increase in methylmalonic acid. Genetic analysis identified a compound heterozygous mutation in the PCCB gene (p.Arg410Trp, c.1228C>T; p.Thr428Ile, c.1283C>T). These mutations had been reported to be frequent among Japanese patients with propionic acidemia;2) thus, the patient was diagnosed with propionic acidemia.
For the treatment of cardiomyopathy, a low-protein diet and L-carnitine were added to enalapril, carvedilol, and levetiracetam. At 1-year examination after the initiation of treatment, she was in good physical condition without worsening of cardiac function.
In previous reports, inborn errors of metabolism account for 4–11.5% of DCM in children,3–5) however, the proportion of propionic acidemia among patients with DCM caused by congenital metabolic disorders is unknown. DCM associated with propionic acidemia usually presents within the first year of life;6) hence, adult-onset DCM with propionic acidemia tends to be unrecognized. This patient did not develop metabolic crisis in the neonatal period, and although she had mild mental retardation and epilepsy, there was no obvious episode of metabolic decompensation. Therefore, DCM was noticed by chance in the absence of any subjective symptoms. Blood acylcarnitine and urinary organic acid analyses revealed that DCM was due to propionic acidemia. Developmental delay, movement disorders/dystonia, and seizures are chronic symptoms of the nervous system in propionic acidemia.1) Therefore, following the development of cardiomyopathy in a patient with developmental delay or seizures, inborn errors of metabolism (e.g., propionic acidemia) may be a differential diagnosis.
The pathophysiology of cardiomyopathy in propionic acidemia is not fully understood. Romano et al. reported that the development of DCM is not associated with age at the time of diagnosis of propionic acidemia, the frequency of metabolic decompensation, or the residual activity of PCC.7) Therefore, clinicians should bear in mind that such a milder form of propionic acidemia like the present case can present with late-onset DCM.
In the present case, the increase in mitochondria in the myocardium, and enlargement of the myocardial mitochondria with swirling cristae were observed under electron microscopy. Kölker et al. pointed out the possibility of mitochondrial damage as pathogenesis of cardiomyopathy in propionic acidemia.8) The changes of mitochondria in the myocardium observed in the present case may be the findings supporting this hypothesis.
In most patients with propionic acidemia, L-carnitine is administered to compensate for secondary carnitine deficiency caused by urinary loss of carnitine bound to organic acids.1) Several investigators reported that the levels of carnitine in cardiac tissue were low, despite the normal levels of plasma free carnitine. In the present case, at least, the level of her plasma free carnitine was normal, however, we did not measure the levels of carnitine in the cardiac tissue. Mardach et al. reported that, in an autopsied case with propionic acidemia who expired due to hypertrophic cardiomyopathy and ventricular fibrillation, the levels of total and free carnitine in myocardial tissue were low despite having long-term carnitine replacement and the normal level of plasma free carnitine.9) Although the mechanism responsible for the low levels of carnitine in the myocardium remains unclear, it may be involved in the development of cardiomyopathy in patients with propionic acidemia.
A few reports have discussed the pathological findings of cardiomyopathy in propionic acidemia. Some of those did not find obvious abnormalities,6, 10) while others showed cardiomyocyte hypertrophy,11) lymphocytes infiltration,12) or localized fibrosis.7) Currently, there is no consensus regarding the specific findings for cardiomyopathy in propionic acidemia. In the present case, hypertrophy of cardiomyocytes and PAS-positive vacuoles were observed around the nuclei of some cardiomyocytes. The vacuoles were negative for PAS-diastase staining, suggesting that these vacuoles contained glycogen. We could not find previous reports performing PAS staining for a biopsied cardiac specimen from patients with propionic acidemia. It is not possible to determine whether these findings are specific to DCM in patients with propionic acidemia. Further study about pathological findings of cardiomyopathy in propionic acidemia may be needed to elucidate whether the deposition of glycogen on the myocardium is specific for this condition.
In conclusion, congenital metabolic disorders, such as propionic acidemia, can be a cause of cardiomyopathy. Physicians should consider screening for inborn errors of metabolism in patients with unexplained cardiomyopathy.
謝辞Acknowledgments
We would like to thank Dr. Go Tajima for genetic diagnosis of propionic acidemia. We wish to thank Prof. Atsushi Manabe for his thoughtful contribution.
Funding
This research was supported by AMED under Grant Number JP17ek0109276.
Conflicts of Interest
All authors have no conflict of interest.
Ethical Approval
Informed consent was obtained from the patient’s parents.
引用文献References
1) Baumgartner MR, Hörster F, Dionisi-Vici C, et al: Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 2014; 9: 130
2) Yang X, Sakamoto O, Matsubara Y, et al: Mutation spectrum of the PCCA and PCCB genes in Japanese patients with propionic acidemia. Mol Genet Metab 2004; 81: 335–342
3) Towbin JA, Lowe AM, Colan SD, et al: Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006; 296: 1867–1876
4) Cox GF: Diagnostic approaches to pediatric cardiomyopathy of metabolic genetic etiologies and their relation to therapy. Prog Pediatr Cardiol 2007; 24: 15–25
5) Miranda JO, Costa L, Rodrigues E, et al: Paediatric dilated cardiomyopathy: Clinical profile and outcome. The experience of a tertiary centre for paediatric cardiology. Cardiol Young 2015; 25: 333–337
6) Riemersma M, Hazebroek MR, Helderman-van den Enden ATJM, et al: Propionic acidemia as a cause of adult-onset dilated cardiomyopathy. Eur J Hum Genet 2017; 25: 1195–1201
7) Romano S, Valayannopoulos V, Touati G, et al: Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J Pediatr 2010; 156: 128–134
8) Kölker S, Burgard P, Sauer SW, et al: Current concepts in organic acidurias: Understanding intra- and extracerebral disease manifestation. J Inherit Metab Dis 2013; 36: 635–644
9) Mardach R, Verity MA, Cederbaum SD: Clinical, pathological, and biochemical studies in a patient with propionic acidemia and fatal cardiomyopathy. Mol Genet Metab 2005; 85: 286–290
10) Ameloot K, Vlasselaers D, Dupont M, et al: Left ventricular assist device as bridge to liver transplantation in a patient with propionic acidemia and cardiogenic shock. J Pediatr 2011; 158: 866–867, author reply, 867
11) Lee TM, Addonizio LJ, Barshop BA, et al: Unusual presentation of propionic acidemia as isolated cardiomyopathy. J Inherit Metab Dis 2009; 32 Suppl 1: S97–S101
12) Laemmle A, Balmer C, Doell C, et al: Propionic acidemia in a previously healthy adolescent with acute onset of dilated cardiomyopathy. Eur J Pediatr 2014; 173: 971–974
 1,Hiromi Kanno-Okada2,Hayato Aoyagi3Yasuhiro Ueda1, Atsuhito Takeda
1,Hiromi Kanno-Okada2,Hayato Aoyagi3Yasuhiro Ueda1, Atsuhito Takeda