Case 1
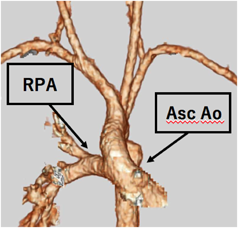
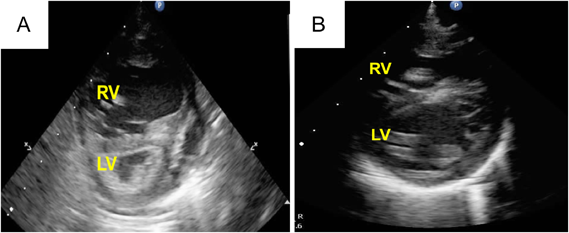
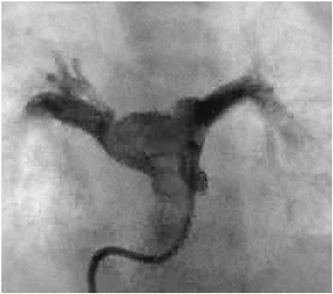
A 25-day-old girl with a complicating RS virus infection was transferred to our hospital because of respiratory distress. Enhanced computed tomography (CT) showed that the right PA originated from the posterolateral wall of the ascending aorta (Fig. 1). Echocardiography revealed that the right ventricle (RV) was remarkably dilated, with a flattened interventricular septum (Fig. 2A) and moderate tricuspid regurgitation (TR) with a pressure gradient of 68 mmHg, suggesting excessive PH. Echocardiography also showed a 5.7-mm-wide atrial septal defect (ASD) with bidirectional shunting. Due to the RS virus infection and requirement for ventilator support, right PAB was planned for initial palliation to avoid cardiopulmonary bypass.
At 36 days of age, right PAB was performed through a median sternotomy using a 2-mm-wide expanded polytetrafluoroethylene (ePTFE) tape. The right PA was gradually tightened to a circumference of 14.5 mm with the band. At that time, the peak blood flow velocity across the right PA was 4.2 m/s. The systemic arterial pressure was finally elevated to 20 mmHg under 100% oxygen and nitric oxide (NO) inhalation. After this surgery, we were able to start aggressive PH therapy using tadalafil and macitentan to improve RV function. This patient was extubated on postoperative day (POD) 24, and NO inhalation was discontinued on POD 32. Echocardiography showed that the deformity of the intraventricular septum had improved, and the TR was trivial with a pressure gradient of 42 mmHg (Fig. 2B). Postoperative cardiac catheterization after right PAB demonstrated that the left pulmonary vascular resistance (PVR) was still high at 10.7 units·m2. However, her left lung reacted well to oxygen supplementation, and the patient was therefore considered to be a good candidate for total correction. After the development of bacterial pneumonia, her cardiac function and PH deteriorated, and mechanical ventilation was required on POD 45. After improvement of the PH and heart failure by antibiotic therapy, total correction was achieved 72 days after the initial palliation.
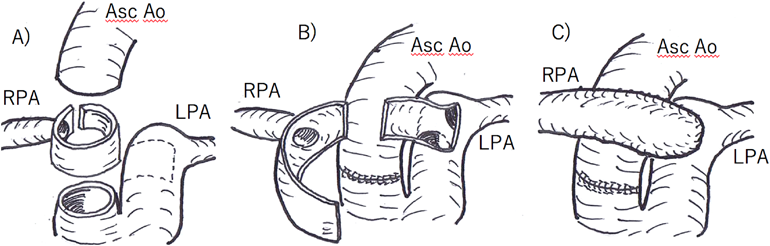
After a repeat sternotomy, cardiopulmonary bypass was established using the innominate artery with a 3.5-mm ePTFE graft and bicaval cannulation. The operative schema for right PA reconstruction is depicted in Fig. 3. This right PA reconstruction was performed with the heart beating. The patient underwent extubation and cessation of NO inhalation on POD 8. Echocardiography showed improvement of the intraventricular septal deformity. Enhanced CT 1 month after the total correction showed no stenosis in the repaired right PA. Postoperative cardiac catheterization 2 months after the total correction demonstrated an MPA pressure of 35/15 (21) mmHg, calculated PVR of 3.43 units·m2, and pulmonary-to-systemic arterial pressure ratio of 0.34. No significant pressure gradient was noted between the RV and both PAs. The patient was discharged on POD 58 with administration of tadalafil and macitentan. One year after surgery, she was asymptomatic with a good general condition.
Case 2
A 1-month-old girl was transferred to our hospital because of AORPA, which had been diagnosed by echocardiography in another hospital. Echocardiography revealed that the RV was remarkably dilated, with a flattened interventricular septum and severe TR with pressure gradient of 97 mmHg. The right PA originated from the ascending aorta. Echocardiography also revealed a small ASD with bidirectional shunting. Enhanced CT showed that the right PA originated from the posterolateral wall of the ascending aorta. Preoperative cardiac catheterization demonstrated a pulmonary arterial pressure of 76/27 (52) mmHg and calculated PVR of 10.83 units·m2. Because of severe PH and a poor reaction to oxygen loading, we hesitated to perform primary total correction. Instead, we decided that right PAB should be performed for initial palliation.
On hospitalization day 7, right PAB was performed through a median sternotomy using a 1.5-mm-wide tape made of an ePTFE graft. The banding tape encircling the right PA was tightened to a circumference of 15 mm. At that time, the peak velocity across the right PA was 3.6 m/s. The systemic arterial pressure was elevated under 100% oxygen and NO inhalation, and intraoperative transesophageal echocardiography showed that the TR and RV function had improved. After right PAB, echocardiography showed only mild TR and an ASD with dominant left-to-right shunting. The RV function further improved with administration of sildenafil, oxygen therapy, and NO inhalation. Although the patient was extubated on POD 2, she required reintubation because of deterioration of the PH due to atelectasis on POD 4. After stabilization of her respiratory condition, total correction was achieved 13 days after right PAB.
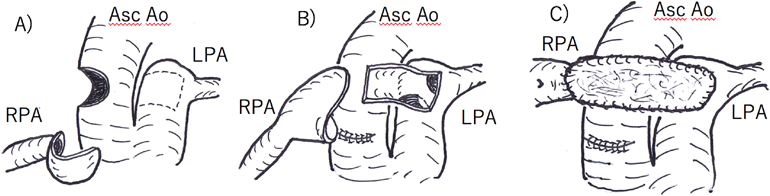
Fig. 4 shows the operative findings in Case 2. Right PA plasty was performed using almost the same procedure as in Case 1 except for the use of patch augmentation with fresh autologous pericardium for right PA reconstruction because of concern about stretching the right PA. After right PA augmentation, the RV-to-systemic artery pressure ratio decreased to 0.55. The patient was was extubated on POD 3, and her postoperative course was uneventful. Postoperative cardiac catheterization 20 days after the total correction showed no stenosis in the reconstructed right PA (Fig. 5). The pulmonary arterial pressure was normalized to 20/7 (10) mmHg, and the calculated PVR was 2.8 units·m2 (Table 1).
Table 1 Postoperative cardiac catheter data in Case 2 | Pressure (mmHg) | RpI (units·m2) | Pp/Ps |
|---|
| LPA (s/d/m) | 20/7 (10) | 2.80 | 0.17 |
| RPA (s/d/m) | 19/4 (10) |
The patient was discharged on POD 25 with administration of sildenafil and macitentan. Five months after surgery, she was asymptomatic and in a good general condition.
AORAP was first described by Fraentzel in 18683) and classified into two subgroups according to the morphological features: a proximal and distal type. In the proximal form, the anomalous PA originates from the right posterior aspect of the proximal ascending aorta close to the aortic valve. About 85% of AORPAs are regarded as the proximal form according to the report by Kutsche & Van Mierop.4) In contrast, in the distal type, the anomalous PA originates from the ascending aorta just proximal to the innominate artery or from the base of the artery itself. In patients with the distal type, the anomalous RPA has a narrow site that is usually distant from its origin.5) The most common anomaly associated with AORPA is a patent ductus arteriosus, followed by an aortopulmonary septal defect, interrupted aortic arch, patent foramen ovale, ventricular septal defect, ASD, coarctation of the aorta, and tetralogy of Fallot. No associated anomalies are found in approximately 15% of patients.6) The natural history of AORPA is characterized by the onset of severe pulmonary vascular obstructive disease, and the prognosis is dismal without an early operation.7) However, it is generally recognized that the early diagnosis and prompt surgical treatment of AORPA results in excellent early and late outcomes. Nathan et al.3) reported that the probability of freedom from death at 20 years was 94%. Therefore, surgical treatment is recommended as soon as a diagnosis is confirmed during the neonatal period.
Armer et al.8) reported the first successful surgical correction of AORPA with interposition of a Dacron graft between the right PA and the main PA in 1961. Kirkpatrick et al.9) performed the first anatomical correction using only autologous tissue. In 1996, van Son and Hanley7) described aortic and main PA flaps for repair of AORPA, thereby reducing tension on the right PA–main PA anastomosis and decreasing the risk of kinking or stenosis of either branch of the PA. Furthermore, in 2002, Prifti et al.10) described a double surgical flap technique. Other modifications of aortic and pulmonary arterial flap techniques have also been described.11) In our two patients, we successfully performed an aortic and pulmonary arterial flap technique using only autologous tissue for the right PA reconstruction. The ascending aorta, which was removed with the flap for the right PA and reanastomosed by itself, tended to be shorter and move posteriorly. There is some concern about narrowing the space behind the ascending aorta. Therefore, the right PA was reconstructed anterior to the ascending aorta. This reconstruction procedure was almost the same as the Lecompte maneuver for the arterial switch operation. However, in the case of transposition of the great arteries, the great arteries exhibit an anterior–posterior relationship; i.e., the PA is positioned anterior to the aorta. Conversely, in the normal side-by-side relationship in these cases, such techniques may cause aortic compression against the reconstructed right PA. The new right PA coursed over the ascending aorta and moved down posteriorly immediately behind the superior vena cava. Therefore, we performed the right PA augmentation with an autologous pericardial patch in Case 2.
The indication and surgical procedure for unilateral PAB has not been well established because unilateral PAB has been rarely performed. In our experience of treating newborns and young infants, we tighten the right PA to a 14- to 15-mm banding circumference with a 3.5- to 4.0-m/s peak velocity across the band on intraoperative epicardial echocardiography.
Few reports have described staged repair of AORPA. In Case 1, right PAB was initially performed, and total correction was delayed because cardiac surgery with cardiopulmonary bypass in patients with symptomatic RS virus infection is associated with a high risk of postoperative PH.2) In Case 2, we hesitated to perform primary total correction because preoperative cardiac catheterization demonstrated a PVR of >10 units·m2, and the patient did not have a good reaction to oxygen loading. Kido et al.12) performed staged repair of AORPA due to severe PH and severely impaired RV function. However, the indication for staged repair of AORPA in patients with severely depressed RV function and PH has not yet been established. Although primary total correction in neonates is the treatment of choice for AORPA, it is important to estimate the reactiveness of PVR against oxygen loading by cardiac catheterization in cases such as those described herein.
Whereas excessive pulmonary arterial blood flow causes hypertension in the anomalous PA, the blood pressure of the contralateral PA from the pulmonary arterial trunk is elevated, sometimes over the systemic arterial pressure, because the entire cardiac output is received from the RV. The advantages of right PAB for AORPA are 1) the avoidance of cardiopulmonary bypass and 2) the ability to start NO inhalation, oxygen therapy, and administration of a pulmonary vasodilator for PH of the left lung. In our patients, TR and RV contraction improved after right PAB. However, complicating pneumonia or atelectasis may cause the deterioration of left PH after right PAB. In fact, both of our patients required reintubation because of deterioration of PH due to bacterial pneumonia in Case 1 and atelectasis in Case 2. In summary, long-term management of AORPA after right PAB is difficult. Therefore, once stabilization of the patient’s general condition has been confirmed, total correction should be performed as soon as possible.