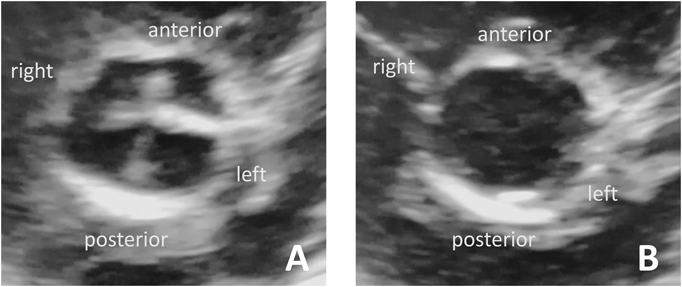
A girl weighing 2,988 g was born without asphyxia at a gestational age of 38 weeks by scheduled cesarean section as her mother previously underwent cesarean section. Her mother was administered levothyroxine for well-controlled hypothyroidism; however, her father and two siblings were healthy. After birth, the patient developed tachypnea with mild hypoxia. No cardiac murmur was noted on auscultation. Although the ductus arteriosus demonstrated physiological patency on echocardiography, no other critical heart anomalies were identified. A diagnosis of transient tachypnea of the newborn was established using chest X-ray. The patient was then admitted to the neonatal intensive care unit of our institute and was administered low-concentration oxygen. Her respiratory impairment rapidly improved, and oxygen administration was discontinued at the age of 1 day. Thereafter, her condition was uneventful, except for the ongoing phototherapy for hyperbilirubinemia. Although echocardiography revealed a completely closed ductus arteriosus at the age of 6 days, a quadricuspid aortic valve (QAV) was incidentally detected (Fig. 1A, B). It was difficult to accurately determine the type of QAV using transthoracic echocardiography alone because the cusps of the aortic valve seemed to differ only slightly in size, and there were no extremely small cusps. QAV of type A, C, or E based on the Hurwitz and Roberts classification was considered to be most likely. The thickened coaptation line between the left anterior and left posterior leaflets was noticeable during diastole (Fig. 1A). Furthermore, the mobility of the commissure seemed to be slightly restricted in systole (movie), suggesting some deformities of the leaflets and commissure. However, color and pulse Doppler echocardiography showed neither valvular regurgitation (AR) nor a stenotic flow pattern, such as turbulent flow. Unfortunately, the coronary anomalies were not evaluated in detail because we did not focus on them. The patient grew well until the age of 1 month, when she was referred to another institute for her regular heart checkup, which revealed a systolic heart murmur. Echocardiography revealed mild pulmonary stenosis with a tricuspid pulmonary valve.
QAV is an extremely rare, congenital cardiac defect marked by four cusps of varying size. According to a large echocardiography database at the Mayo Clinic, it occurs in 0.006% of individuals.1) Fewer cases have been reported in children because isolated QAV without other cardiac defects is generally asymptomatic in childhood. Although the cause of this aortic valvular dysmorphogenesis remains unclear, an in vitro study revealed that the redundant cusp may result from the division of one of the three mesenchymal anlagens to mature into a normal aortic valve.2)
QAVs are classified into seven types according to the size and distribution of the leaflets. Type A has four equal-sized cusps; type B has three equal-sized cusps and one small-sized cusp; type C has two equal-sized, large cusps and two equal-sized, small cusps; type D has one large-sized cusp, two intermediate-sized cusps, and one small-sized cusp; type E has three equal-sized cusps and one large-sized cusp; type F has two equal-sized, large cusps and two equal-sized, small cusps; and type G has four unequal-sized cusps. Types A, B, and C account for 80% of all types of QAVs. Although QAV usually occurs as an isolated defect, it can be associated with other malformations, such as patent ductus arteriosus, ventricular septal defect, pulmonary stenosis, tetralogy of Fallot, and coronary anomalies.1)
QAV can be complicated by AR, valvular stenosis, aortic dilatation, and infective endocarditis. AR, the most predominant and principal complication, tends to progress at over 40 years of age. Progressive cusp fibrosis is considered as the primary mechanism underlying subsequent coaptation failure. According to data from the Mayo Clinic, 90% of patients with QAV presented with AR (of any degree); of these patients, 26% had moderate or severe AR, and there was no association between QAV subtype and the severity of regurgitation. Reportedly, the location of the accessory cusp is not well correlated with the occurrence of AR.3) The most common, conventional, surgical treatment method of dysfunctional QAV is prosthetic valve replacement. Recently, there have been reports of successful aortic valve repair, such as tricuspidization or bicuspidization.4, 5) Tricuspidization is the most common method of repair for the QAVs of types A, B, and C.4) In addition, a successful bicuspidization has been reported for the QAV of type G.5) In the present case, it was impossible to predict the degree of severity of valve dysfunction and the best method of surgical treatment in adulthood. A tailor-made approach corresponding to various pathologic morphologies of the aortic valve is expected under the direct and detailed observation of the surgeon. Coronary anomalies should be accurately evaluated prior to surgery because of the possibility of concomitant repair. Although some patients may require surgical intervention for symptomatic AR, QAV generally has a good life prognosis, characterized by the slow progression of AR and the survival of patients similar to that of healthy individuals matched for age and sex.1)
In the present case, fortunately, QAV was incidentally detected at an early age. If this diagnosis was not established, then AR and other complications may have developed without symptoms in late middle age. The patient will require regular checkups for cardiovascular monitoring throughout her life.
Currently, echocardiography is essential for the evaluation of hemodynamics in neonatal care. We should focus on the morphology of the semilunar valve and should ensure that it is evaluated as a part of routine echocardiography at least once.
Conflicts of Interest
The authors have no conflicts of interest to declare.
Note
The supplementary movie of this article can be viewed online.
引用文献References
1) Tsang MYC, Abudiab MM, Ammash NM, et al: Qudricuspid aortic valve: Characteristics, associated structural cardiovascular abnormalities and clinical outcomes. Circulation 2016; 133: 312–319
2) Fernández B, Durán AC, López AMD, et al: New embryological evidence for the formation of quadricuspid aortic valves in the Syrian hamster (Mesocricetus auratus). J Comp Pathol 1999; 121: 89–94
3) Naito K, Ohteki H, Yunoki J, et al: Aortic valve repair for quadricuspid aortic valve associated with aortic regurgitation and ascending aortic aneurysm. J Thorac Cardiovasc Surg 2004; 128: 759–760
4) Williams L, Peters P, Shah P: Tricuspidization of qudricupid aortic valve. Ann Thorac Surg 2013; 95: 1453–1455
5) Luciani GB, Morjan M, Faggian G, et al: Repair of quadricuspid aortic valve by bicuspidization: A novel technique. Interact Cardiovasc Thorac Surg 2010; 11: 348–350