Mitochondrial Cardiomyopathy
Atsuhito Takeda Atsuhito Takeda
Atsuhito Takeda
Atsuhito TakedaDepartment of Pediatrics, Faculty of Medicine and Graduate School of Medicine, Hokkaido University ◇ Hokkaido, Japan
発行日:2020年7月1日Published: July 1, 2020
Mitochondrial cardiomyopathy is characterized by an oxidative phosphorylation (OXPHOS) disorder due to genetic mutations in genes encoding the structure and function of myocardial mitochondria. Mutations in both mitochondrial and nuclear DNA can cause mitochondrial cardiomyopathy, and it is typically recognized as one of the generalized manifestations of neurological and metabolic disorders. Cardiomyopathy, however, can be the only phenotype of mitochondrial disease and is often misdiagnosed. Cardiac manifestations vary from asymptomatic to catastrophic heart failure or sudden death. Although recent evolution in genetic testing has allowed for the identification of the causative gene, tissue sampling to identify an OXPHOS disorder is still regarded as the gold standard for diagnosis.
Key words: mitochondrial cardiomyopathy; oxidative phosphorylation (OXPHOS); mitochondrial DNA; respiratory chain complex
© 2020 特定非営利活動法人日本小児循環器学会© 2020 Japanese Society of Pediatric Cardiology and Cardiac Surgery
Mitochondrial cardiomyopathy is characterized by an oxidative phosphorylation (OXPHOS) disorder due to genetic mutations in genes encoding the structure and function of myocardial mitochondria.1) It is often recognized as one of the phenotypes of common mitochondrial disease, such as MELAS, Leigh encephalopathy, and infantile diseases with muscle weakness or inborn errors of metabolism. However, it is rarely recognized as an isolated mitochondrial cardiomyopathy and is often overlooked when there are no associated symptoms other than heart problems. To date, mitochondrial cardiomyopathy has been diagnosed more accurately and with greater ease by the identification of new causative genes using next-generation sequencers and the functional evaluation method of mitochondria on a cell basis by measuring oxygen consumption rate. Recent studies have also revealed that mitochondria play a major role in failing myocardium, and investigating mitochondrial cardiomyopathy could allow for a better understanding of the genes and enzymes involved in myocardial energy metabolism and also their evaluation systems. In this regard, we will discuss the normal structure and function of mitochondria and the molecular genetics, pathophysiology, clinical features, and definitive diagnosis of mitochondrial cardiomyopathy.
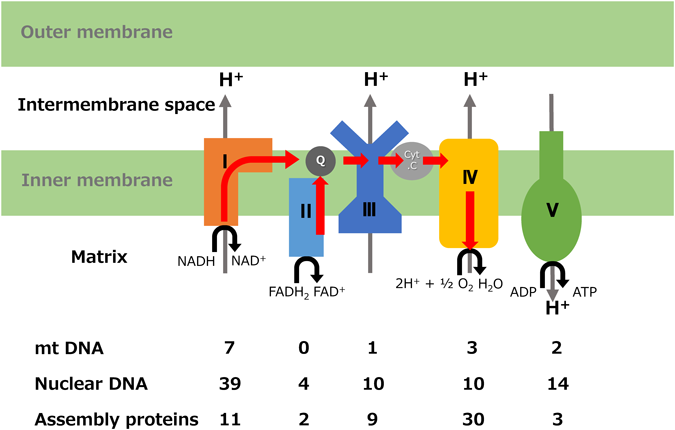
Mitochondria are organelles present in almost all eukaryotic cells that produce ATP by aerobic respiration in tissue on-demand. The heart is an organ that consumes 30 kg of ATP per day in adults, and hundreds of mitochondria exist in one cardiomyocyte. The mitochondria are composed of an outer membrane and an inner membrane, and the inner membrane forms a fold called cristae, which includes an electron transport system composed of four respiratory chain enzyme complexes (RC Complex I to IV) and ATP synthase (Complex V). The inner membrane contains abundant complex lipids called cardiolipin and plays a major role in membrane impermeability and the stabilization of respiratory chain enzyme complexes. NADH and FADH2, which are primary proton and electron carriers, are generated by β-oxidation and the TCA cycle from a substrate such as fatty acid transported to a matrix, and protons are pumped into the intermembrane space by oxidation at the RC. ATP is synthesized in the presence of ATP synthase by a proton gradient across the inner membrane. Simultaneously generated electrons move through the electron transport system (coenzyme Q-Complex III-cytochrome c) and finally reduce oxygen molecules to water (Fig. 1). About 1500 proteins are involved in maintaining the mitochondrial structure and function, but only 1% are encoded by mitochondrial DNA (mtDNA). The remaining 99% are encoded by nuclear genes. mtDNA is a circular DNA of 16569 bases that exists uniquely in mitochondria. Thousands of copies exist in one cardiomyocyte and encode 13 subunits of the RC (except Complex II) as well as 22 tRNAs and 2 rRNAs involved in translation and synthesis2) (Fig. 2). The nuclear gene encodes the remaining RC subunits, assembly factors, quality maintenance of mtDNA, proteins involved in mitochondria transport, etc.; synthesizes the protein in the cytoplasm; and transports it into the mitochondria. The mtDNA replication involves DNA polymerase γ (POLG) and the Twinkle protein that functions as an mtDNA helicase. Transcription and translation of mtDNA involve mitochondrial RNA polymerase (mtRNAP), mitochondrial transcription factor (TFAM), and translation elongation factor (TSFM), all of which are encoded by nuclear genes.
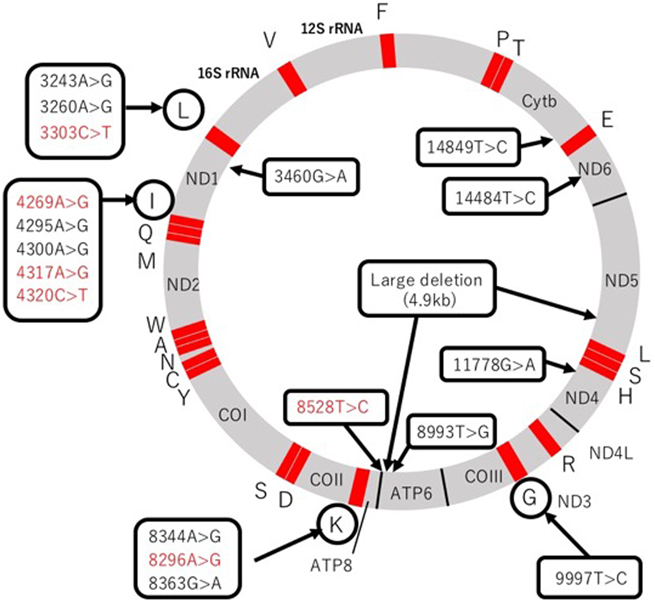
Individual capital letters represent each tRNA. Mutations in red indicate early onset cardiomyopathy during infancy. ND1, ND2, ND3, ND4L, ND4, ND5, ND6: Complex I subunit genes. Cytb: Complex III subunit gene. COI, COII, COIII: Complex IV subunit genes. ATP6, ATP8: Complex V subunit genes.
The causative gene of mitochondrial cardiomyopathy includes mtDNA and nuclear gene mutations, and their genetic backgrounds are very different. However, the cardiac phenotype is similar and could not be differentiated based on the clinical course and symptoms.
There are mtDNA and nuclear genes encoding proteins related to mitochondrial structure and function, but mtDNA is involved only in a few genes related to oxidative phosphorylation (OXPHOS). On the other hand, most mitochondrial diseases are due to nuclear gene mutations. Although there are point mutations and deletions in mtDNA abnormalities, in both cases there is a state in which normal mtDNA and mutant mtDNA are mixed (heteroplasmy), and symptoms occur after exceeding a certain mutation rate (threshold effect). In addition, it is known that this threshold effect does not often develop until adulthood. mtDNA mutation is common in adults, but nuclear gene mutation is also common in children. Moreover, because the mutation rate and threshold vary among the organ or tissue (tissue specificity), it is necessary to consider the mtDNA mutation and mutation rate of the affected organ in order to make a definitive diagnosis of the mtDNA mutation.3) For example, even when the mtDNA mutation rate is very low in the blood, there is a possibility that a high mutation rate of mtDNA exists in the myocardium. In fertilized eggs, mtDNA exists only in oocytes and has a unique form of inheritance called maternal inheritance. Mutation rates vary greatly among siblings, and the age of onset and disease severity vary between families; thus, genetic counseling must be performed.4) Deletion of mtDNA is a characteristic of Kearns-Sayre syndrome, and cardiac conduction disorder is known to occur as a cardiac complication. To date, more than 250 mitochondrial disease-causing genes have been reported in nuclear gene mutations with all inheritance patterns including autosomal dominant inheritance, autosomal recessive inheritance, and X-linked recessive inheritance. In prenatal diagnosis, interpretation of mitochondrial abnormalities of mtDNA mutations is difficult because of heteroplasmy, whereas genetic counseling would be important in lethal diseases because of nuclear gene mutations.
Table 1 shows the genes and phenotypes related to mitochondrial cardiomyopathy.5)
| I. Mitochondrial DNA | Gene | mutation | Cardiomyopathy | Phenotype (OMIM) |
|---|---|---|---|---|
| Subunits of respiratory chain complex | ||||
| MT-ND1 | m.3460G>A | HCM, LVNC | LHON | |
| MT-ND4 | m.11778G>A | HCM | LHON / Progressive Dystonia | |
| MT-ATP6/8 | m.8528T>C | HCM | Infantile cardiomyopathy | |
| MT-ATP6 | m.8993T>G | HCM | NARP / Leigh Disease | |
| MT-ND6 | m.14484T>C | DCM | LHON | |
| MT-CYB | m.14849T>C | HCM | Septo-Optic Dysplasia | |
| Mitochondrial protein synthesis | ||||
| MT-TL1 | m.3243 A>G | HCM, DCM, RCM, LVNC | MELAS / Leigh Syndrome / CPEO / Mitochondrial Mitopathy | |
| m.3260 A>G | HCM, DCM | MELAS/Maternal Myopathy and Cardiomyopathy | ||
| m.3303T>C | HCM, DCM | Maternal Myopathy and Cardiomyopathy | ||
| MT-TI | m.4300 A>G | HCM, DCM | Maternally Inherited Cardiomyopathy | |
| MT-TK | m.8344 A>G | HCM, DCM | MERRF | |
| m.8363G>A | HCM, DCM | MERRF/Leigh Syndrome | ||
| MT-RNR1 | m.1555 A>G | RCM | Maternally inherited DEAFness | |
| II. Nuclear genes | Gene | mutation | Cardiomyopathy | Phenotype (OMIM) |
| Subunits of respiratory chain complex | ||||
| Complex I | NDUFS2 | 252010 | HCM | Mitochondrial complex I deficiency |
| Complex I | NDUFV2 | 252010 | HCM | Mitochondrial complex I deficiency |
| Complex I | NDUFA11 | 252010 | HCM | Mitochondrial complex I deficiency |
| Complex II | SDHA | 252011 | DCM, LVNC | Mitochondrial complex II deficiency |
| Assembly factor | ||||
| Complex I | NDUFAF1 | 252010 | HCM | Mitochondrial complex I deficiency |
| Complex I | ACAD9 | 611126 | HCM | Mitochondrial complex I deficiency due to ACAD9 deficiency |
| Complex IV | SCO2 | 604377 | HCM | CEMCOX1 (fatal infantile cardioencephalomyopathy due to cytochrome c oxidase deficiency 1) |
| Complex IV | COX10 | 220110 | HCM | Mitochondrial complex IV deficiency |
| Complex IV | COX15 | 615119 | HCM | CEMCOX2 (fatal infantile cardioencephalomyopathy due to cytochrome c oxidase deficiency 2) |
| Complex V | TMEM70 | 614052 | HCM | MC5DN2 (mitochondrial complex V (ATP synthase) deficiency nuclear type 2) |
| Mitochondrial protein synthesis | ||||
| AARS2 | 614096 | HCM | COXPD8 (combined oxidative phosphorylation deficiency-8) | |
| MRPS22 | 611719 | HCM | COXPD5 (combined oxidative phosphorylation deficiency-5) | |
| TSFM | 610505 | HCM | COXPD3 (combined oxidative phosphorylation deficiency-3) | |
| GTPBP3 | 616198 | HCM, DCM | COXPD23 (combined oxidative phosphorylation deficiency-23) | |
| MTO1 | 614702 | HCM | COXPD10 (combined oxidative phosphorylation deficiency-10) | |
| ELAC2 | 615440 | HCM | COXPD17 (combined oxidative phosphorylation deficiency-17) | |
| Maintenance of mitochondrial integrity | ||||
| TAZ | 302060 | DCM, LVNC | BTHS (Barth Syndrome) | |
| AGK | 212350 | HCM | Sengers Syndrome | |
| SLC22A5 | 212140 | HCM, DCM | CDSP (Systemic primary carnitine deficiency) | |
| ACADVL | 201475 | HCM, DCM | VLCAD deficiency | |
| HADHA | 609015 | DCM | HADHA (Trifunctional protein deficiency alpha subunit) | |
| Mitochondrial DNA stability | ||||
| SLC25A4 | 615418 | HCM | MTDPS12(mitochondrial DNA depletion syndrome-12) | |
| Iron homeostasis | ||||
| FXN | 229300 | HCM | FRDA1 (Friedreich ataxia) | |
| BOLA3 | 614299 | HCM | MMDS2 (multiple mitochondrial dysfunctions syndrome-2) with hypoglycinemia | |
| Coenzyme Q10 biosynthesis | ||||
| COQ9 | 614654 | HCM | COQ10D5 (coenzyme Q10 deficiency-5) | |
| COQ4 | 616276 | HCM | COQ10D7 (coenzyme Q10 deficiency-7) | |
| Mitochondrial protein transport | ||||
| DNAJC19 | 610198 | DCM, LVNC | MGCA5 (3-methylglutaconic aciduria type V) | |
| DCM; dilated cardiomyopathy, HCM; hypertrophic cardiomyopathy, RCM; restrictive cardiomyopathy, LVNC; left ventricular noncompaction. Modified from Reference 5). | ||||
The functional classification of genes that cause mitochondrial cardiomyopathy is as follows: 1) RC complex subunit, 2) RC complex assembly, 3) mitochondrial protein synthesis, 4) maintenance of mitochondrial integrity, and 5) mitochondrial DNA stability. The mtDNA mutations encoding Complex I, IV, and V subunits6) have been reported as RC complex subunit abnormalities. In nuclear gene mutations, NDUFS2, NDUFV2, and NDUFA11 have been reported as encoding the complex I subunit and cause hypertrophic cardiomyopathy (HCM) with Leigh encephalopathy.7–9) SDHA has been reported in genetic mutations encoding Complex II subunits, which resulted in dilated cardiomyopathy (DCM) and left ventricular non-compaction (LVNC), and is fatal in almost all affected infants.10)
In the assembly factor of the complex I, Leigh encephalopathy is a common disorder, among which NDUFAF1 and ACAD9 are associated with cardiomyopathy.11, 12) SURF1 is famous for the assembly factor of Complex IV, but SCO2, COX10, and COX15 are important as disease-causing genes for cardiomyopathy.13–15) In Complex V, TMEM70 is a factor for early Complex V assembly, and mutations at this site are known to cause infantile lactic acidosis, 3-methylglutaconuria, and cardiomyopathy.16) A gene involved in mitochondrial protein synthesis includes a gene abnormality encoding tRNA in mtDNA, and the m.3243A>G mutation is well known as a causative gene of MELAS.
In nuclear mutations, tRNA post-translational modifications and aminoacyl-tRNA synthetase (aaRS) abnormalities are known, and AARS2 mutations have been reported as a causative gene for infant-onset HCM.17, 18) The gene involved in maintaining mitochondrial integrity is known as the TAZ gene that encodes the enzyme tafazzin involved in cardiolipin maturation, and is known as the responsible gene for Barth syndrome. LVNC is a major cardiac complication in TAZ and is an extremely important gene for differential diagnosis of male infants with severe heart failure.19) Other mitochondrial phospholipid metabolism disorders include mitochondrial disease (Sengers syndrome) characterized by cardiomyopathy and cataract due to acylglycerol kinase deficiency (AGK mutation).20) Among the genes involved in mitochondrial DNA stability, SLC25A4 is known as the causative gene of mtDNA depletion syndrome, and these mutations cause cardiomyopathy.21)
A common pathological condition of mitochondrial cardiomyopathy is a reduced ability to produce ATP in mitochondria per unit myocardium. Myocardium is an organ that continuously consumes energy through aerobic metabolism, and ATP deficiency directly leads to decreased myocardial contractility.22) Fetal myocardium has suppressed aerobic metabolism under a hypoxic environment, and even if mitochondrial dysfunction is present, it may not be serious. After birth, cardiac energy depends on aerobic metabolism by mitochondria, and thus cardiomyopathy develops in neonates and infants if mitochondrial maladaptation exists.23) In mitochondrial cardiomyopathy that develops after infancy, cardiomyocytes are filled with mitochondria as compensation for reduced ATP production, with displaced myofibrils and increased reactive oxygen species.
This may cause further qualitative deterioration of mitochondria and eventually lead to myocardial failure. In clinical practice, it is often recognized as a transition from circumferential left ventricular hypertrophy to dilated phase hypertrophic cardiomyopathy. Other cardiac phenotypes such as LVNC, cardiac conduction disorders, and pulmonary hypertension have been reported, but the detailed mechanisms of these diseases due to mitochondrial dysfunction have not been elucidated.
Mitochondrial cardiomyopathy varies in severity, from asymptomatic to severe heart failure, and sometimes fatal arrhythmias and sudden death may occur.
Biological stress such as infection may worsen the clinical course and even trigger the diagnosis. Mitochondrial cardiomyopathy is usually diagnosed accompanied by associated symptoms such as neuromuscular or metabolic diseases, but cardiomyopathy may be a clue to the diagnosis of mitochondrial disease regardless of the associated symptoms. Typical phenotypes are HCM, DCM, RCM, and LVNC. HCM accounts for about half of mitochondrial cardiomyopathy, most of which is symmetrical left ventricular hypertrophy, but hypertrophic obstructive cardiomyopathy also has been reported. Most HCM would progress from systolic dysfunction to left ventricular decompensation and may shift to dilated phase hypertrophic cardiomyopathy. DCM is the second most common with HCM, and may also occur in KSS, MELAS, MERRF, and Leigh encephalopathy. In cases with isolated cardiomyopathy, complex II deficiency has been reported. A survival rate up to 16 years of age was reported to be 18%,24) and the prognosis is extremely poor. RCM is rare but has a poor prognosis, and 12% of 24 cases of mitochondrial cardiomyopathy undergoing heart transplantation have been reported as RCM.25) Although LVNC is rare in frequency, Finsterer et al. reported that mitochondrial cardiomyopathy had the highest occurrence (40 cases) among 187 cases with left ventricular hypertrabeculation (LVHT)/noncompaction, followed by Barth syndrome (30 cases). Thus, it may be underestimated in practice.26) Histiocytoid cardiomyopathy (Purkinje fiber dysplasia) may develop with ventricular arrhythmia in infancy but has been reported to be one of the mitochondrial diseases.27) Arrhythmia would develop alone or in combination with the above cardiomyopathy. Sinus dysfunction syndrome is found in KSS and mitochondrial DNA depletion syndrome. A complete atrioventricular block is highly associated with KSS, and thus the timing of pacemaker implantation should be noted. WPW syndrome is frequently associated with MELAS and MERRF, and may be indicated for ablation in patients with supraventricular tachycardia.28)
Mitochondrial cardiomyopathy is important as a differential diagnosis of HCM in newborns and infants, and it is necessary to proceed with a definitive diagnosis by the method described below, taking into consideration the possibility of accompanying diseases such as muscle disease, metabolic disease, and hearing loss. Physicians should keep in mind that it may be lethal when complete atrioventricular blocking or ventricular tachycardia occur in MCM. In common mitochondrial diseases (MELAS, MERRF, KSS, LHON, Leigh encephalopathy), cardiomyopathy and arrhythmia may often develop. About 40% of the patients with MELAS have the complication of HCM,29) and there was a positive correlation between the mt.3243A>G mutation rate and the degree of cardiac hypertrophy.30) If it progresses, it often transitions to the dilated phase of HCM. About half of the patients with MERRF are known to have cardiomyopathy such as HCM or DCM.31) In KSS, 45% of the patients had fainting due to an advanced atrioventricular block, and 23% exhibited sudden death. Thus, preventive implantation of a pacemaker should be considered when a fascicular block appears.32) HCM has been reported in LHON and Leigh encephalopathy, but its frequency is unknown.
Fig. 3 shows a flowchart for the definitive diagnosis of mitochondrial cardiomyopathy. A definitive diagnosis of mitochondrial cardiomyopathy, as in the definition above, shows the oxidative phosphorylation disorder in the myocardium and the causative gene. Diagnosis of oxidative phosphorylation disorder is based on Bernier’s diagnostic criteria for mitochondrial respiratory chain disorder (Table 2) and is widely used in neuromuscular diseases.33) According to this diagnostic criteria, it is necessary to measure enzyme activity using myocardium in the case of isolated cardiomyopathy. Also, as noted in the diagnostic criteria, mitochondrial abnormalities of myocardium in an electron microscopic image can also help with the diagnosis. The diagnostic criteria for mitochondrial cardiomyopathy in electron microscopy has not been defined yet. However, abnormal mitochondrial proliferation, degeneration of cristae (concentric circles, bundles), and accumulation of glycogen granules are considered suspicious findings. Light microscopy often shows vacuolar degeneration. In autopsy cases such as sudden death of unknown cause, it is useful to measure respiratory chain enzyme activity in the myocardium when cardiomyopathy is suspected. Note that in both biopsy and autopsy, the collected tissue should be kept untreated and stored frozen at −80 degrees to preserve enzyme activity. Formalin (formaldehyde) solution is a fixative used only for light microscopy, but light microscopic images have poor specific findings for mitochondrial cardiomyopathy, and a definitive diagnosis is not possible if all the extracted heart tissue is immersed in formalin. For electron microscopy, it is important to first immerse in glutaraldehyde solution. If the tissue is not fixed for electron microscopy, it may be possible to proceed to the preparation of an electron microscope sample at the formalin fixation stage, but the preparation from a paraffin-embedded block results in a significantly degraded sample and pathologic diagnosis becomes difficult. In newborns and infants, cardiac biopsy is not performed from the first stage of causative diagnosis for cardiomyopathy, and if there are accompanying symptoms of mitochondrial disease, genetic testing will be performed. If no abnormalities are found in a common genetic test for mitochondrial disease, a whole-exome search using a next-generation sequencer is performed. Recently, genetic diagnosis using a genetic panel for mitochondrial disease with nuclear genes and mitochondrial DNA has been utilized for a quick and efficient solution (Fig. 4). If there are skeletal muscle symptoms such as muscle weakness, skeletal muscle biopsy is also available. Fig. 5 shows a case of mitochondrial cardiomyopathy diagnosed by skeletal muscle biopsy and endomyocardial biopsy. The individual was found to have mild muscle weakness in the differential diagnosis of HCM, which became an indicator of possible mitochondrial disease. In this case, a definitive diagnosis was made using the two major criteria of Ragged-red fiber in skeletal muscle and decreased respiratory chain enzyme activity in the heart muscle. Recently, the oxygen consumption rate of fibroblasts from skin biopsies has emerged as a method of the diagnosis of oxidative phosphorylation disorder.34) Genetic diagnosis is very important, but it is limited because there are so many types of related genes. In the future, it is necessary to improve the accuracy of diagnosis with low-invasive modalities, especially genetic testing. Thus, it is extremely important to establish a registration system and a medical network for such a rare disease.
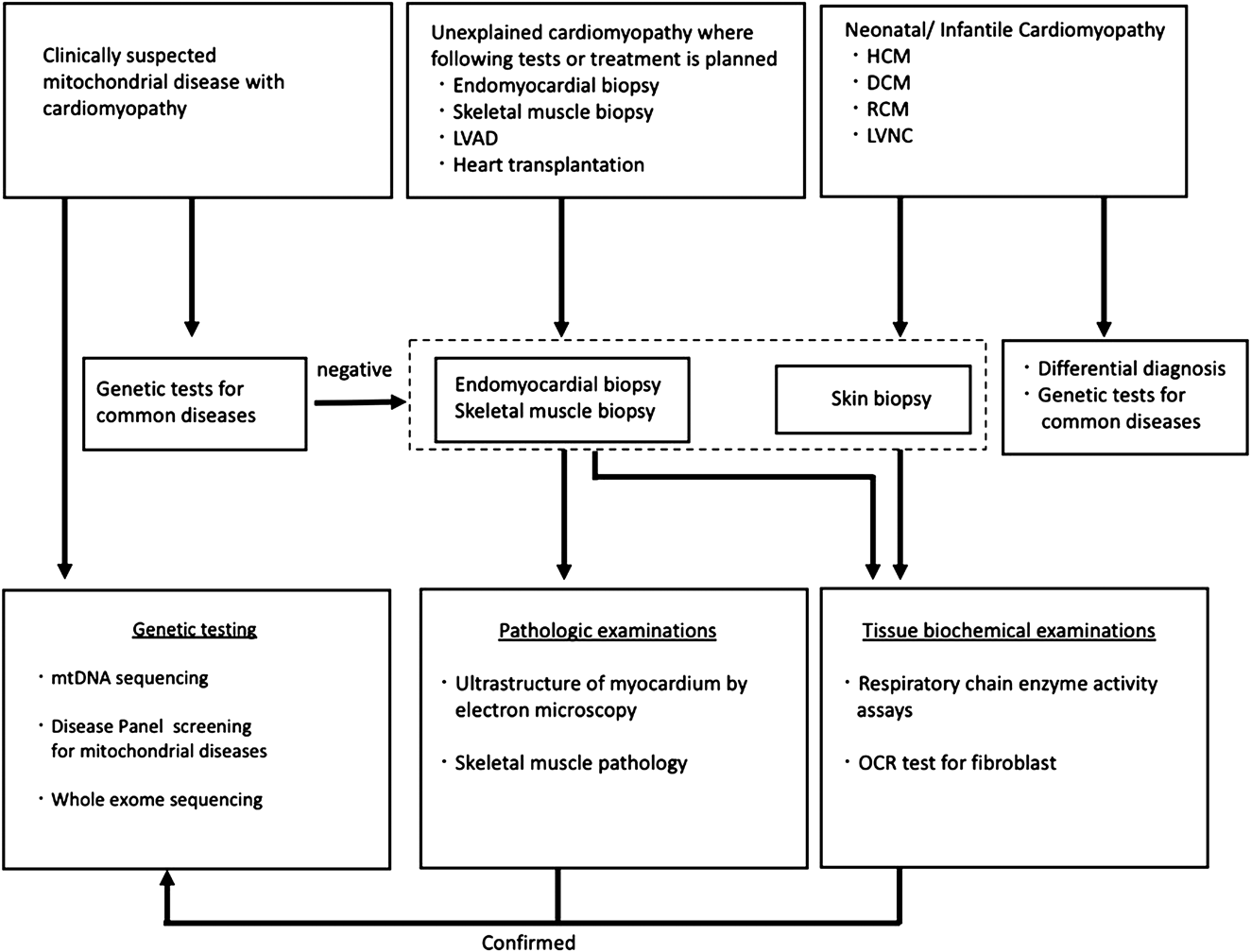
Genetic tests for common mitochondrial diseases: MELAS (mt3243A>G, mt3271T>C), MERRF (mt8344A>G), Leigh encephalopathy (mt8993T>G), KSS/CPEO(mtDNA deletion). Differential diagnosis: Pompe disease, Noonan syndrome, organic acidemias and fatty acid oxidation disorders, sarcomere gene mutations, and Barth syndrome (TAZ). Modified from Reference 5).
| ■Major diagnostic criteria |
| I. Clinical |
| Clinically complete RC encephalomyopathy or a mitochondrial cytopathy defined as fulfilling all three of the following conditions |
| 1. Unexplained combination of multisystemic symptoms that is essentially pathognomonic for a RC disorder. |
| Symptoms must include at least three of the organ system presentations described elsewhere, namely neurologic, muscular, cardiac, renal, nutritional, hepatic, endocrine, hematologic, otologic, ophthalmologic, dermatologic, or dysmorphic. |
| 2. A progressive clinical course with episodes of exacerbation (e.g., following intercurrent illnesses) or a family history that is strongly indicative of a mtDNA mutation (at least one maternal relative other than the proband whose presentation predicts a probable or definite RC disorder). |
| 3. Other possible metabolic or nonmetabolic disorders have been excluded by appropriate testing, which may include metabolite, enzyme, or mutation analyses, imaging, electrophysiological studies, and histology. |
| II. Histology |
| > 2% ragged red fibers in skeletal muscle |
| III. Enzymology |
| >2% COX-negative fibers if <50 years of age |
| >5% COX-negative fibers if >50 years of age |
| <20% activity of any RC complex in a tissue |
| <30% activity of any RC complex in a cell line |
| <30% activity of the same RC complex activity in two tissues |
| IV. Functional |
| Fibroblast ATP synthesis rates >3SD below mean |
| V. Molecular |
| Identification of a nuclear or mtDNA mutation of undisputed pathogenicity |
| ■Minor diagnostic criteria |
| I. Clinical |
| Symptoms compatible with a RC defect |
| II. Histology |
| 1%–2% ragged red fibers if aged 30–50 years |
| Any ragged red fibers if <30 years of age |
| >2% subsarcolemmal mitochondrial accumulations in a patient<16 years of age |
| Widespread electron microscopic abnormalities in any tissue |
| III. Enzymology |
| Antibody-based demonstration of a defect in RC complex expression |
| 20%–30% activity of any RC complex in a tissue |
| 30%–40% activity of any RC complex in a cell line |
| 30%–40% activity of the same RC complex activity in two tissues |
| IV. Functional |
| Fibroblast ATP synthesis rates 2–3 SD below mean |
| Fibroblasts unable to grow on media with glucose replaced by galactose |
| V. Molecular |
| Identification of a nuclear or mtDNA mutation of probable pathogenicity |
| VI. Metabolic |
| One or more metabolic indicators of impaired RC function |
| 1. High levels of lactate, pyruvate and alanine in blood and cerebrospinal fluid |
| 2. Increased protein in cerebrospinal fluid (when KSS is suspected) |
| 3. Abnormal findings in 31P-MRS or PET (muscle or brain) |
| 4. Abnormal findings in ergometer (decrease in VO2max, AVO2D, lactate threshold) |
| Definite diagnosis: two major criteria or one major plus two minor criteria. |
| Probable diagnosis: one major plus one minor criterion or at least three minor criteria. |
| Possible diagnosis: a single major criterion or two minor criteria, one of which must be clinical. |
| Modified from Reference 33). |

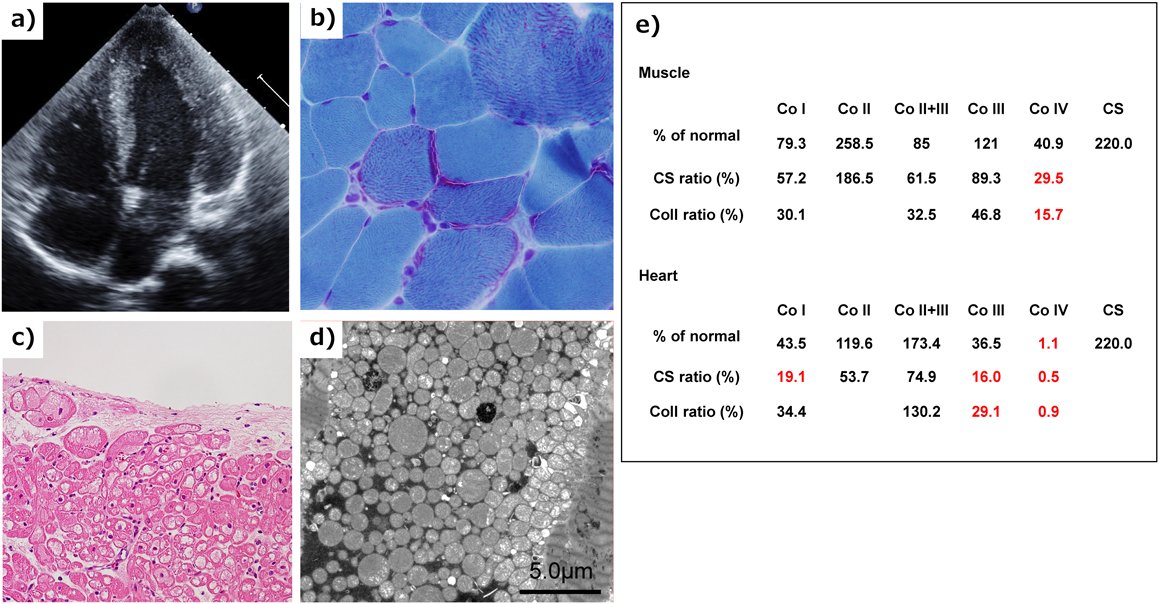
a) Echocardiography showed mild hypertrophic cardiomyopathy. b) Skeletal muscle biopsy with modified Gomori trichrome staining showed ragged-red fibers. c) Light microscopy of a biopsied right ventricle showed cytoplasmic vacuolization in the myocardium. d) Electron microscopy of a biopsied right ventricular endomyocardium showed marked proliferation of mitochondria within myofibrils. e) Respiratory chain complex activity of biopsied skeletal muscle and myocardium showed defects in complex IV and combined defects in Complex I, III, and IV. Co I, Co II, Co II+III, Co III, Co IV: Enzymatic activity of respiratory chain complex in complex I, II, II+III, III, and IV. CS: citrate synthase activity.
There is no established treatment for mitochondrial cardiomyopathy, and general medical management should be performed according to chronic heart failure. Renin-angiotensin inhibitors and beta-blockers should be considered for cases with cardiac dysfunction such as dilated cardiomyopathy or dilated phase hypertrophic cardiomyopathy, but the prognostic effect is unknown. Renin-angiotensin inhibitors should be used with caution when there is renal dysfunction. For acute exacerbation of heart failure caused by mitochondrial cytopathy, intravenous administration of diuretics and PDE-III inhibitors are used to improve congestion in addition to vitamin cocktail therapy. It may develop with fatal arrhythmias such as ventricular tachycardia and ventricular fibrillation. When it occurs, amiodarone and group Ia drugs should be considered after cardioversion. Prediction of fatal arrythmia before onset is difficult in patients with mitochondrial cardiomyopathy, so it is not indicated for ICD implantation as primary prevention of sudden death, but is indicated for secondary prevention. For refractory severe heart failure, ventricular assist device (VAD) treatment is also considered when heart transplantation is indicated. However, mitochondrial disease is often associated with other organ involvement, and the indication of heart transplantation should be carefully considered in view of the severity of the effects in other organs.
From 2015, clinical research has been developed to establish clinical guidelines, a diagnostic system, and a registry system to improve the quality of medical treatment of mitochondrial disease in Japan. The registry system is also being developed in the MO-Bank (Mitochondrial disease research Organization data Bank) (HP: http://mo-bank.com/index.html) and in cooperation with a Global Mito Registry, an international registry system for mitochondrial diseases.
Finally, there is currently no specific treatment for mitochondrial cardiomyopathy, and traditional cardioprotective and symptomatic treatments are being performed. It is inevitable that the treatment differs depending on the causative gene, and the greatest difficulty is that the drug does not easily pass through the inner membrane of the mitochondria. However, in the course of disease progression, there are common pathways, such as mitochondrial disorders associated with an increase in reactive oxygen species, and it is likely that they will be targets for treatment in the future. In addition, research on drug delivery to the mitochondria has recently emerged, and we hope that a major paradigm shift will occur in the future.
We appreciate Dr. Kei Murayama (Department of Metabolism, Chiba Children’s Hospital, Chiba, Japan) who provided data on respiratory chain enzyme activity, Dr. Koji Okazaki(Diagnostics and Therapeutics of Intractable Diseases, Intractable Disease Research Center, Graduate School of Medicine, Juntendo University, Tokyo, Japan), Dr. Akira Ohtake (Department of Pediatrics & Clinical Genomics, Faculty of Medicine, Saitama Medical University, Saitama, Japan, Center for Intractable Diseases, Saitama Medical University Hospital, Saitama, Japan), who provided the gene panel for mitochondrial disease, and Dr. Akira Sudo (Nirenokai Children’s Clinic) for providing the pathology information for skeletal muscles.
There is no conflicts of interest to disclosure.
Originally published in Pediatric Cardiology and Cardiac Surgery, Vol. 33(2017), No. 4, pp. 287–296
1) Meyers DE, Basha HI, Koenig MK: Mitochondrial cardiomyopathy: Pathophysiology, diagnosis, and management. Tex Heart Inst J 2013; 40: 385–394
2) Schapira AH: Mitochondrial disease. Lancet 2006; 368: 70–82
3) Rossignol R, Faustin B, Rocher C, et al: Mitochondrial threshold effects. Biochem J 2003; 370: 751–762
4) Vento JM, Pappa B: Genetic counseling in mitochondrial disease. Neurotherapeutics 2013; 10: 243–250
5) Murayama K, Osaka H, Yonada M: Clinical manual for mitochondrial diseases 2017. Shindan to Chiryo-sha 2016: 69–76
6) Imai A, Fujita S, Kishita Y, et al: Rapidly progressive infantile cardiomyopathy with mitochondrial respiratory chain complex V deficiency due to loss of ATPase 6 and 8 protein. Int J Cardiol 2016; 207: 203–205
7) Loeffen J, Elpeleg O, Smeitink J, et al: Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann Neurol 2001; 49: 195–201
8) Benit P, Beugnot R, Chretien D, et al: Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum Mutat 2003; 21: 582–586
9) Berger I, Hershkovitz E, Shaag A, et al: Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann Neurol 2008; 63: 405–408
10) Levitas A, Muhammad E, Harel G, et al: Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur J Hum Genet 2010; 18: 1160–1165
11) Fassone E, Taanman JW, Hargreaves IP, et al: Mutations in the mitochondrial complex I assembly factor NDUFAF1 cause fatal infantile hypertrophic cardiomyopathy. J Med Genet 2011; 48: 691–697
12) Leslie N, Wang X, Peng Y, et al: Neonatal multiorgan failure due to ACAD9 mutation and complex I deficiency with mitochondrial hyperplasia in liver, cardiac myocytes, skeletal muscle, and renal tubules. Hum Pathol 2016; 49: 27–32
13) Leary SC, Mattman A, Wai T, et al: A hemizygous SCO2 mutation in an early onset rapidly progressive, fatal cardiomyopathy. Mol Genet Metab 2006; 89: 129–133
14) Antonicka H, Leary SC, Guercin GH, et al: Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum Mol Genet 2003; 12: 2693–2702
15) Antonicka H, Mattman A, Carlson CG, et al: Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am J Hum Genet 2003; 72: 101–114
16) Diodato D, Invernizzi F, Lamantea E, et al: Common and Novel TMEM70 Mutations in a Cohort of Italian Patients with Mitochondrial Encephalocardiomyopathy. JIMD Rep 2015; 15: 71–78
17) Mazurova S, Magner M, Kucerova-Vidrova V, et al: Thymidine kinase 2 and alanyl-tRNA synthetase 2 deficiencies cause lethal mitochondrial cardiomyopathy: Case reports and review of the literature. Cardiol Young 2017; 27: 936–944
18) Gotz A, Tyynismaa H, Euro L, et al: Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet 2011; 88: 635–642
19) Takeda A, Sudo A, Yamada M, et al: Eponym: Barth syndrome. Eur J Pediatr 2011; 170: 1365–1367
20) Siriwardena K, Mackay N, Levandovskiy V, et al: Mitochondrial citrate synthase crystals: Novel finding in Sengers syndrome caused by acylglycerol kinase (AGK) mutations. Mol Genet Metab 2013; 108: 40–50
21) Strauss KA, DuBiner L, Simon M, et al: Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc Natl Acad Sci USA 2013; 110: 3453–3458
22) El-Hattab AW, Scaglia F: Mitochondrial cardiomyopathies. Front Cardiovasc Med 2016; 3: 25
23) Marin-Garcia J, Goldenthal MJ: The mitochondrial organelle and the heart. Rev Esp Cardiol 2002; 55: 1293–1310
24) Scaglia F, Towbin JA, Craigen WJ, et al: Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 2004; 114: 925–931
25) Bates MG, Nesbitt V, Kirk R, et al: Mitochondrial respiratory chain disease in children undergoing cardiac transplantation: A prospective study. Int J Cardiol 2012; 155: 305–306
26) Finsterer J: Cardiogenetics, neurogenetics, and pathogenetics of left ventricular hypertrabeculation/noncompaction. Pediatr Cardiol 2009; 30: 659–681
27) Finsterer J: Histiocytoid cardiomyopathy: A mitochondrial disorder. Clin Cardiol 2008; 31: 225–227
28) Finsterer J, Kothari S: Cardiac manifestations of primary mitochondrial disorders. Int J Cardiol 2014; 177: 754–763
29) Anan R, Nakagawa M, Miyata M, et al: Cardiac involvement in mitochondrial diseases: A study on 17 patients with documented mitochondrial DNA defects. Circulation 1995; 91: 955–961
30) Majamaa-Voltti K, Peuhkurinen K, Kortelainen ML, et al: Cardiac abnormalities in patients with mitochondrial DNA mutation 3243A>G. BMC Cardiovasc Disord 2002; 2: 12
31) Catteruccia M, Sauchelli D, Della Marca G, et al: “Myo-cardiomyopathy” is commonly associated with the A8344G “MERRF” mutation. J Neurol 2015; 262: 701–710
32) Barrera-Ramirez CF, Barragan-Campos HM, Ilarraza H, et al: Cardiac involvement in Kearns-Sayre syndrome. Rev Esp Cardiol 2005; 58: 443–446
33) Bernier FP, Boneh A, Dennett X, et al: Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002; 59: 1406–1411
34) Ogawa E, Shimura M, Fushimi T, et al: Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: A study of 106 Japanese patients. J Inherit Metab Dis 2017; 40: 685–693
This page was created on 2020-06-23T17:00:14.686+09:00
This page was last modified on 2020-07-17T15:06:59.000+09:00
このサイトは(株)国際文献社によって運用されています。
