Noonan syndrome with multiple lentigines (NSML), previously called LEOPARD (Lentigines, Café au lait spots, Electrocardiographic abnormalities, Pulmonary stenosis, Obstructive cardiomyopathy, Abnormalities of genitalia, Ocular hypertelorism, Retarded growth, Deafness, Skeletal abnormalities) syndrome, is a rare autosomal dominant inherited disorder with congenital malformations caused by abnormal intercellular RAS/mitogen activated protein kinase (RAS/MAPK) signaling. Clinical manifestations of NSML often overlap with those of Noonan syndrome, Costello syndrome, and cardiofaciocutaneous syndrome, all of which are caused by the genetic abnormalities of proteins involved in the RAS/MAPK signaling system. Therefore, these diseases are often referred to as RASopathies.1) Common cardiac complications of NSML include electrocardiogram abnormalities and hypertrophic cardiomyopathy (HCM). HCM has been reported in 80% of NSML cases, 40% of which have severe left ventricular outflow tract obstruction (LVOTO).2) Standard treatments for HCM are often ineffective and can lead to sudden death.3, 4) Therefore, there is a need for the development of an effective treatment. NSML is caused by mutations in the PTPN11, RAF1, and BRAF genes, which are the components of the RAS/MAPK signaling system. Among them, the frequency of abnormalities is particularly high in the PTPN11 gene, which encodes the SH2 domain-containing protein tyrosine phosphatase 2 (SHP2). Approximately 90% of patients with NSML have heterozygous missense mutations in the PTPN11 gene.5) Clinical manifestations of RASopathies, including NSML, can result from the excess signaling of the RAS/MAPK cascade. However, recently, SHP2 has been shown to be important not only for the RAS/MAPK cascade, but also for correct signaling through the AKT/mammalian target of rapamycin (AKT/mTOR) pathway.5, 6) In addition, it was reported that HCM in mice harboring the heterozygous missense variant of c. 836A>G (p.Y279C) in the PTPN11 gene was secondary to AKT/mTOR hyperactivity, and the administration of the mTOR inhibitor, rapamycin, reverses cardiac hypertrophy.7) Although mTOR inhibitors have been shown to improve HCM associated with NSML mouse model, there are few reports on its clinical experience. Herein, we report our experience with the use of an mTOR inhibitor, a rapamycin analog, as a treatment for a patient with NSML complicated by HCM with severe LVOTO.
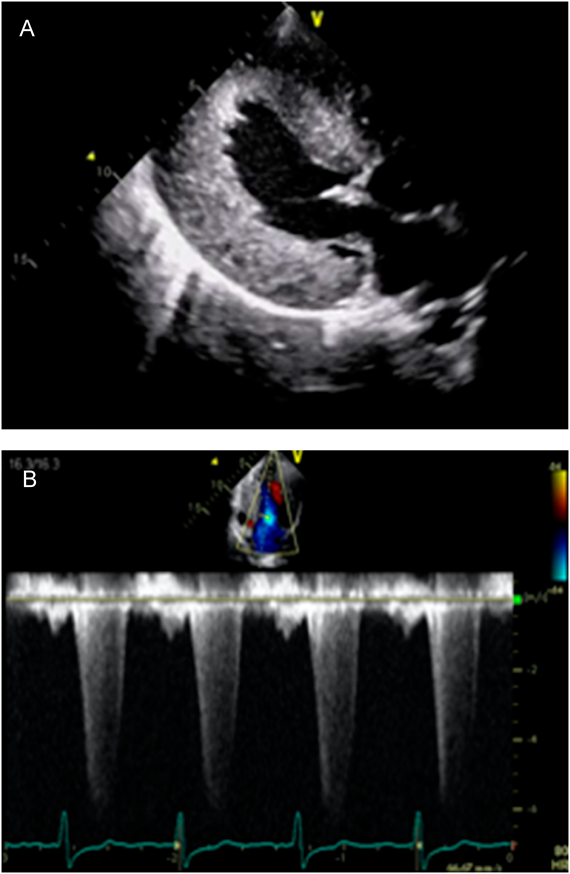
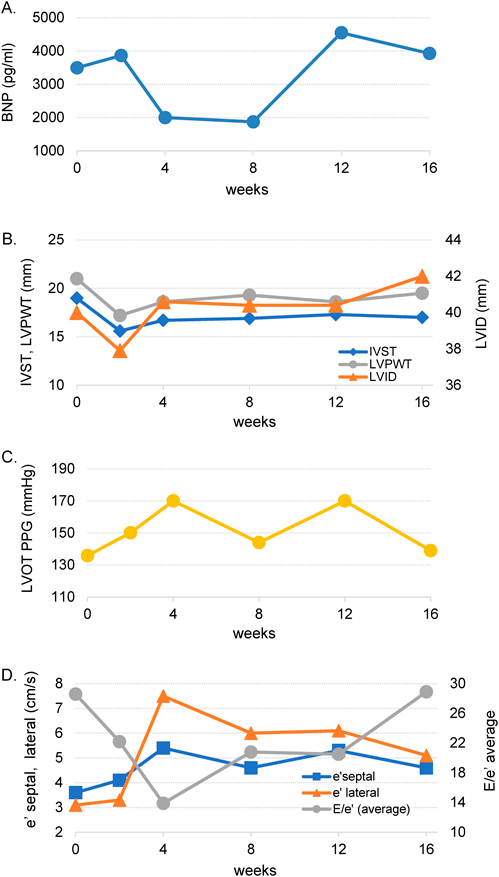
The patient is a 19-year-old Japanese man. He was born to non-consanguineous parents at the gestational age of 38 weeks. At the age of 2 days, a heart murmur was observed. He was transferred to our hospital for further investigation. Clinical examination revealed facial dysmorphism with widely spaced eyes and low-set ears. We performed echocardiography, which revealed significantly thickened left ventricular posterior wall (LVPW) and interventricular septal thickness (IVST). The measurements of IVST and LVPW at the end-diastolic phase were 12.2 and 11.9 mm, respectively. Severe LVOTO was also noted with a peak pressure gradient of 93 mmHg. We diagnosed the patient as having Noonan syndrome based on clinical manifestations. LVOTO was severe, however, there were no symptoms of heart failure; therefore, the patient was managed regularly on an outpatient basis. Although the patient had no subjective symptoms, LVOTO remained severe. We started propranolol administration when the patient was 5 years old. However, there was no significant change in the echocardiography parameter. His lentigines were not particularly noticeable in early childhood, but the number of lentigines increased with his growth. Based on clinical symptoms, we suspected the patient of having NSML. To confirm, we conducted a genetic testing at the age of 12 years and observed a heterozygous missense mutation NM_001330437.2: c.1528C>G (p.Q510E) in exon 13 of the PTPN11 gene that was previously reported in patients with NSML.8) The patient had no complaints and was able to live his daily life without any symptoms. However, if this condition is left untreated, sudden death from a fatal arrhythmia may occur. Surgical intervention had to be considered, however, both parents and patient were reluctant to perform highly invasive surgical treatment. Therefore, we decided to perform treatment using an mTOR inhibitor, rapamycin analog, sirolimus. We initiated sirolimus treatment when the patient was 19 years old. Treatment began after approval by the ethics committee of St. Marianna University (IRB approval #3522), and a written informed consent was obtained from his parents. The dose of sirolimus was determined with reference to the dose used for lymphangioleiomyomatosis and was scheduled to be taken daily (1 mg/day) for 16 weeks. To confirm the efficacy of the treatment, blood test, electrocardiogram, chest X-ray, and echocardiography were performed 2, 4, 8, 12, and 16 weeks before and after sirolimus administration. Echocardiography performed before sirolimus treatment showed significantly thickened entire left ventricular wall. IVST, left ventricular internal dimension (LVID), and LVPWT at the end-diastolic phase were 19, 40, and 21 mm, respectively. The calculated peak pressure gradient of LVOTO was 136 mmHg. In tissue Doppler method, both e′ septal and e′ lateral were slightly low with 3.6 cm/s and 3.1 cm/s, respectively, and the calculated average E/e′ was as high as 28.6, which suggested a decrease in the LV diastolic function and an increase in the left atrial pressure (Fig. 1A, B). Chest X-ray examination showed cardiomegaly with a cardiothoracic ratio of 60% without pulmonary congestion. Electrocardiographic findings were suggestive of severe left ventricular hypertrophy and myocardial damage, such as left axis deviation, high RV5 potential, and negative T wave at V4–6. Brain natriuretic peptide (BNP) level was significantly elevated to 3,495.6 pg/mL. Other blood test data were within the normal range. The echocardiographic measurements of the end-diastolic phase, IVST, LVD, LPWT, peak pressure gradient of LVOT, tissue Doppler measurements of e′ septal, e′ lateral, and E/e′, and BNP changes are shown in Fig. 2A–D. BNP level decreased after administering sirolimus; however, the data at 16 weeks after the start of administration were almost equal to the data before administration. No major changes were observed in the values measured using echocardiography and tissue Doppler method.
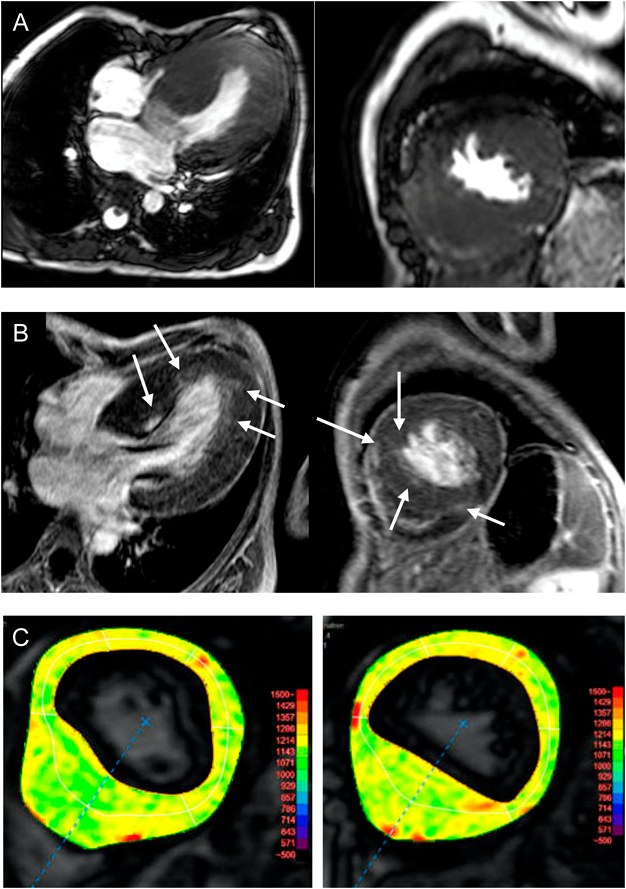
The blood trough concentration of sirolimus in the 16th week after administration was 6.0 ng/mL (effective range, 5–15 ng/mL); therefore, the dose was considered appropriate. Stomatitis occurred during sirolimus administration but did not require special treatment. There were no other side effects that could be attributed to sirolimus administration. Eight weeks after the completion of sirolimus treatment, cMRI was performed (Fig. 3A–C). Significant wall thickening of the entire left ventricle was confirmed using cine MRI. Late gadolinium enhancement (LGE) examination revealed patchy gadolinium enhanced spots in the basal and apical area. T1 mapping revealed elevated native T1 value in the entire left ventricle, suggesting that fibrosis occurred in the entire left ventricle. Echocardiography performed at the same time as the cardiac MRI examination 8 weeks after the end of the treatment showed significant thickened IVS and LVPW with 18 mm each, significant high E/e′ (average) with 24.8, and peak PG of LVOT 137.5 mmHg. No obvious changes could be confirmed from the measured values before treatment. The patient subsequently underwent myectomy because of the limited availability of other medical treatment. The patient’s subsequent course was stable.
We administered sirolimus for 16 weeks to a patient with NSML with severe LVOTO, however, no obvious effect could be confirmed. T1 mapping on cardiac MRI confirmed an elevation in T1 value throughout the left ventricle, suggesting that myocardial fibrosis occurred in the entire left ventricle. Myocardial fibrosis is a histological change that occurs in the end stages of heart failure regardless of the cardiac disease. The effect of rapamycin is expected to be exerted by suppressing cell proliferation caused by RASopathy. The effect on the myocardium where fibrosis occurs extensively as in this patient cannot be expected. Therefore, the results may have been different if rapamycin was started before myocardial fibrosis occurred.
The clinical manifestations of RASopathies are caused by the abnormal RAS/MAPK signaling, but crosstalk occurs between the RAS/MAPK and PI3K/AKT signaling for balancing each other9); therefore, PI3K/AKT system is also associated with the clinical manifestations of RASopathies. Approximately 90% of NSML is caused by a genetic mutation in the PTPN11 gene, which encodes SHP2.5) SHP2 has an important role not only in the RAS/MAPK pathway but also in the AKT/mTOR pathway.5, 6) In addition, Martin et al. reported that HCM in NSML mice models harboring the heterozygous PTPN11 Y279C mutation was secondary to AKT/mTOR hyperactivity.7) Moreover, it was revealed that hypertrophic cardiomyopathy was ameliorated by early administration of rapamycin. It has been reported that an mTOR inhibitor such as rapamycin may be a therapeutic agent for HCM associated with NSML; however, there are few reports on its clinical experience.
Hahn et al. have reported their experience in using a rapamycin analog for treating HCM in NSML infants with a heterozygous Q510 mutation in the PTPN11 gene.10) Although the patient was to undergo a heart transplant eventually, rapamycin treatment had improved the New York Heart Association classification stage and decreased the BNP levels. Based on these findings, it was concluded that rapamycin may be effective for HCM associated with NSML.10) In our case, we could not confirm the changes of measurement values and parameter of cardiac function which was measured by tissue Doppler methods on echocardiography associated with the sirolimus treatment. However, a few points merit discussion: Post-treatment cardiac MRI results suggested that T1 mapping showed elevated T1 levels throughout the left ventricle, suggesting fibrosis occurred throughout the entire left ventricle. However, in LGE, only spot-like contrast findings were observed, and there was a gap between the two methods. While traditional LGE techniques could detect regional variations in myocardial contrast enhancement compared to the normal myocardium, accurate determination of variations becomes difficult when the myocardium is affected widely.11) In addition, it has been reported that about half of HCM patients do not have LGE positive findings.12)
Native T1 mapping is an MRI technique that measures the absolute T1 value of the myocardium. T1 mapping can accurately evaluate myocardial properties even in cardiomyopathy that involves the entire left ventricle.13) Fibrosis of the left ventricular myocardium is caused by a long-term pressure overload. Moreover, myocardial fibrosis was not normalized by the current treatment. Considering the MRI findings, we probably initiated the treatment after the onset of advanced fibrosis rendering the treatment less effective, and the results may have been different if the treatment was initiated at an earlier stage. Furthermore, the RAS/MAPK and AKT/mTOR signaling systems are both complex pathways; even if the signaling system downstream of mTOR is suppressed by an mTOR inhibitor, signals may be mediated through other pathways. Therefore, proliferation or cell hypertrophy may not be completely suppressed with sirolimus treatment alone, and it might be necessary to simultaneously suppress proteins involved in cell growth and differentiation in the AKT/mTOR and RAS/MAPK systems. Addition of agents such as GSK-3β and ERK1/2 to the treatment regimen may be warranted for better treatment of the condition. When should we initiate rapamycin administration? Scharmm et al. have published an interesting study that provides insight on this.14) In their studies of HCM mice models using loss-of-function mutation Q510-SHP2, mice have shown increased cardiomyocyte size, heart-to-body weight ratio and interventricular septum dimeter and cardiomyocyte disarray from the prenatal period. They have also reported that interstitial fibrosis occurred in a 3-month-old heart. Interestingly, these changes could be suppressed when rapamycin was administered early after birth. Based on their findings, rapamycin may be a drug that should be given as early as possible, rather than when general treatment is ineffective. Although the effect of rapamycin could not be confirmed in our case at this time, its effect on HCM associated with NSML cannot be completely ruled out. Further studies are warranted, including the careful selection of cases such as initiating therapy at an earlier stage and/or addition of other agents to the treatment regimen.
Abbreviations
BNP, brain natriuretic peptide; HCM, hypertrophic cardiomyopathy; IVST, interventricular septum thickness; LGE, late gadolinium enhancement; LVID, left ventricular internal dimension; LVOTO, left ventricular outflow obstruction; LVPW, left ventricular posterior wall; mTOR, mammalian target of rapamycin; NSML, noonan syndrome with multiple lentigines
Funding
No funding was secured for this study.
Conflicts of Interest
The authors report no conflicts of interest.
Contributors’ Statement
Dr. Aso designed the study and drafted the initial manuscript. Dr. Sakurai, Dr. Masumori, Dr. Nakano, and Dr. Osada collected and analyzed the patient data. Dr. Migita performed and interpreted the genetic test and Dr. Kotoku performed and interpreted the cardiac MRI. All the authors have reviewed and revised the manuscript and have approved the final manuscript for submission and agree to be accountable for all aspects of the work.
引用文献References
1) Tidyman WE, Rauen KA: The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 2009; 19: 230–236
2) Limongelli G, Pacileo G, Marino B, et al: Prevalence and clinical significance of cardiovascular abnormalities in patients with the LEOPARD syndrome. Am J Cardiol 2007; 100: 736–741
3) Woywodt A, Welzel J, Haase H, et al: Cardiomyopathic lentiginosis/LEOPARD syndrome presenting as sudden cardiac arrest. Chest 1998; 113: 1415–1417
4) Limongelli G, Sarkozy A, Pacileo G, et al: Genotype-phenotype analysis and natural history of left ventricular hypertrophy in LEOPARD syndrome. Am J Med Genet A 2008; 46: 620–628
5) Gelb BD, Tartaglia M: RAS signaling pathway mutations and hypertrophic cardiomyopathy: Getting into and out of the thick of it. J Clin Invest 2011; 121: 844–847
6) Digilio MC, Lepri F, Baban A, et al: RASopathies: Clinical diagnosis in the first year of life. Mol Syndromol 2011; 1: 282–289
7) Marin TM, Keith K, Davies B, et al: Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 Mutation. J Clin Invest 2011; 121: 1026–1043
8) Digilo MC, Sarkozy A, Pacileo G, et al: PTPN11 gene mutations: Linking the Gln510Glu mutation to the “LEOPARD syndrome phenotype”. Eur J Pediatr 2006; 165: 803–805
9) Aksamitiene E, Kiyatkin A, Kholodenko BN: Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: A fine balance. Biochem Soc Trans 2012; 40: 139–146
10) Hahn A, Lauriol J, Thul J, et al: Rapidly progressive hypertrophic cardiomyopathy in an infant with Noonan syndrome with multiple lentigines: Palliative treatment with a rapamycin analog. Am J Med Genet A 2015; 167A: 744–751
11) Kim RJ, Chen EL, Lima JAC, et al: Myocardial Gd-DTPA kinetics determine MRI contrast enhancement and reflect the extent and severity of myocardial injury after acute reperfused infarction. Circulation 1996; 94: 3318–3326
12) Todiere G, Aquaro GD, Piaggi P, et al: Progression of myocardial fibrosis assessed with cardiac magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol 2012; 60: 922–929
13) Lu M, Zhao S, Yin G, et al: T1 mapping for detection of left ventricular myocardial fibrosis in hypertrophic cardiomyopathy: A preliminary study. Eur J Radiol 2013; 82: 225–231
14) Schramm C, Fine DM, Edwards MA, et al: The PTPN11 loss-of-function mutation Q510E-Shp2 causes hypertrophic cardiomyopathy by dysregulating mTOR signaling. Am J Physiol Heart Circ Physiol 2012; 302: H231–H243
 1,Kenzo Sakurai1,Yosuke Osada1,Marie Nakano1,Chikako Masumori1,Ohsuke Migita1,Akiyuki Kotoku2Kentaro Aso
1,Kenzo Sakurai1,Yosuke Osada1,Marie Nakano1,Chikako Masumori1,Ohsuke Migita1,Akiyuki Kotoku2Kentaro Aso