Pediatric heritable pulmonary arterial hypertension (HPAH) with ALK-1 (ACVRL-1) mutation is usually known to be severe and progressive, compared to pediatric idiopathic pulmonary arterial hypertension (IPAH) or HPAH with BMPR2 mutation.1, 2) There is no previous report of pediatric patients with ALK-1 mutation associated with HPAH, who improved without recent pulmonary vasodilators.
We present here, a case of pediatric HPAH carrying an ALK-1 mutation who was initially diagnosed as severe primary pulmonary hypertension but was improved without disease specific therapy.
In the early 1990’s, a six-year-old boy was admitted to our hospital for an episode of syncope on exercise at his elementary school. He had frequent nasal bleeding. Although the distance from his home to his elementary school was only 3-minutes on foot, he could not walk this distance because of fatigue. There was no familial history of pulmonary arterial hypertension.
His S2 heart sound seemed accentuated. Electrocardiogram showed marked right ventricular hypertrophy and the echocardiogram revealed his right ventricular systolic pressure (RVSP) to be as high as 100 mmHg without any congenital heart defect. Cardiac catheterization revealed his main pulmonary arterial pressure was 89/39 (65) mmHg, and pulmonary vascular resistance index (PVRI) was 14.7 Wood unit m2 (Table 1). Acute vasoreactivity test (AVT) with inhaled oxygen and intravenous tolazoline did not decrease his pulmonary arterial pressure and PVRI. The six-minute walk test (6MWT) was not performed because of fatigue. Plain computed tomography of his chest showed no specific finding and lung ventilation and perfusion scintigraphy showed normal. Autoimmune antibodies were all negative.
Table 1 Cardiac catheterization at the age of 6 years | Room air | O2 inhalation | NO inhalation |
|---|
| RA | 6 | | |
| RV | 87/8 | | |
| PA | 89/39(65) | 98/50(74) | 112/71(91) |
| PAWP | 8 | | |
| LV | 119/19 | | |
| AO | 102/58(79) | 99/45(66) | 111/54(74) |
| Cl (L/min/m2) | 3.96 | | 5.79 |
| PVR (Wood unit×m2) | 14.38 | | 13.47 |
| PVR/SVR | 0.78 | | 1.05 |
| SaO2 (%) | 97 | | |
| NO: nitric oxide, RA: right atrium, RV: right ventricle, PA: pulmonary artery, PAWP: pulmonary arterial wedge pressure, LV: left ventricle, AO: aorta, CI: cardiac index, PVR: pulmonary vascular resistance, PVR/SVR: ratio of pulmonary vascular resistance per systemic vascular resistance |
We diagnosed him as severe primary pulmonary hypertension (PPH). Back then, there was no established effective medication for PPH and home oxygen therapy was refused by his parents. Although we suggested him for limitation of exercises and started him on bunazosin hydrochloride (alpha-1 adrenergic receptor blocker which was considered to be effective for PPH at that time), he was unable to take the medication regularly due to headache, abdominal pain, and dizziness.
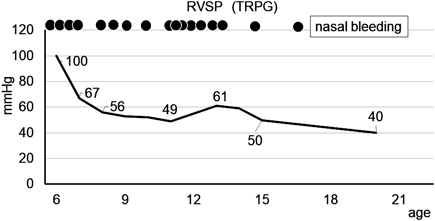
Despite of insufficient therapies for PAH in the outpatient clinic, he showed few symptoms and his RVSP measured from the tricuspid regurgitation flow velocity on echocardiogram showed a decrease(Fig. 1). Later in junior high school, he had no difficulty in physical activity and participated in all the school activities including exercise. At the age of 13, RVSP on echocardiogram was high at 61 mmHg and we started him on beraprost, but again, he could not take it regularly due to the minor adverse effects. At the age of 14, we discontinued bunazosin and beraprost since he was irregular with taking the medications. At the age of 20 years, his RVSP on echocardiogram decreased to 40 mmHg; subsequently we could not evaluate his RVSP because his tricuspid regurgitation flow became too small to measure. He discontinued follow-up because he felt no symptom and no difficulty in his daily university life.
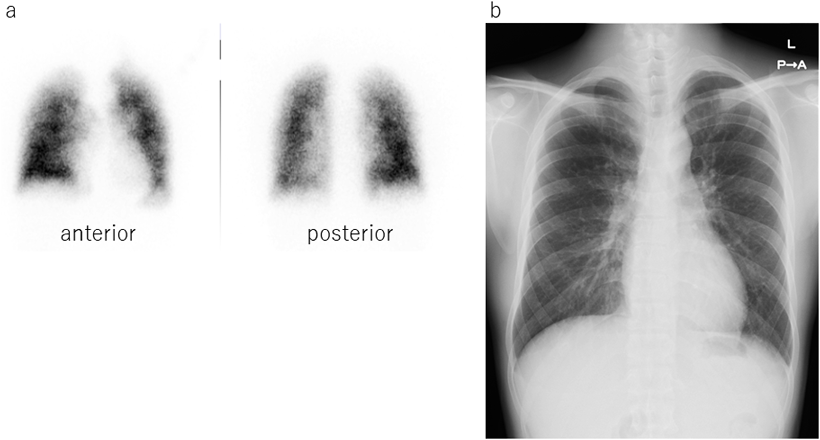
At the age of 28 years, he visited our outpatient clinic again because he recognized the severity and the poor prognosis of PAH through his university curriculum. He had no symptoms in his daily work and could easily go upstairs to 5 floors at his office. He could walk over 660 m in 6MWT and SpO2 were slightly decreased from 98% to 93% after 6MWT. His serum brain natriuretic protein was as low as below normal level. Echocardiogram revealed his right ventricle was not dilated and had preserved contraction. Chest X-ray showed mild dilatation on pulmonary trunk and lung perfusion scintigraphy showed mild diffuse patchy pattern which was typical in PAH(Fig. 2). Chest plain computed tomography could not detect abnormality. Treadmill cardiopulmonary functional test revealed SpO2 depletion from 98% to 92%.
He had still frequent nasal bleeding. There was one spot of telangiectasia on his left leg. We suspected hereditary hemorrhagic telangiectasia (HHT) and suggested follow up cardiac catheterization and genetic testing for ALK-1 gene, along with BMPR2 and ENG.
The cardiac catheterization revealed his main pulmonary arterial pressure had reduced to 51/18(32) mmHg, PVRI was 7.8 Wood unit m2 but AVT to oxygen and nitric oxide inhalation was still negative (Table 2).
Table 2 Cardiac catheterization at the age of 28 years | Room air | O2 inhalation | NO inhalation |
|---|
| RA (mmHg) | 5 | | |
| RV | 48/7 | | |
| PA | 50/18(32) | 50/18(30) | 51/18(33) |
| PAW | 8 | | |
| LV | 99/11 | | |
| AO | 98/61(78) | 92/53(74) | 100/63(75) |
| Cl (L/min/m2) | 2.65 | 3.18 | 3.05 |
| PVR (Wood unit×m2) | 7.83 | 6.28 | 7.21 |
| PVR/SVR | 0.36 | 0.29 | 0.31 |
| SaO2 (%) | 99 | | |
| NO: nitric oxide, RA: right atrium, RV: right ventricle, PA: pulmonary artery, PAWP: pulmonary arterial wedge pressure, LV: left ventricle, AO: aorta, CI: cardiac index, PVR: pulmonary vascular resistance, PVR/SVR: ratio of pulmonary vascular resistance per systemic vascular resistance |
The genetic testing revealed he carried a novel ALK-1 missense mutation (p.Leu193Pro (c.578T>C)). No pathogenic variant was identified in BMPR2 or ENG. The Leu193Pro variant of ALK-1 gene is located in a highly conserved GS domain of ALK-1 and classified as “Likely Pathogenic” in ClinVar (ID 548119). We diagnosed him as HPAH.
Although his pulmonary arterial pressure and PVRI were still high compared to normal level, they decreased without any treatment and he had maintained exercise capacity and no clinical signs of heart failure. After discussion about his treatment, we concluded that pulmonary vaso-dilatator therapy was not indicated and establishing regular follow up was necessary for him. After 3 years since catheterization, he still had no exercise limitation and worked without any symptom. Echocardiogram showed the same findings compared to those measured at the catheterization, and serum BNP level was not elevated.
According to the United States National Institute of Healthcare registry, IPAH and HPAH were grave disorders with poor prognosis in the early 1990’s, since no effective therapy was known then.3) Although several effective pulmonary vasodilators have been discovered in the last 15 years, the survival rate of pediatric HPAH is still unsatisfactory, according to the REVEAL registry.4) In the French registry, HPAH with ALK-1 mutation also had a poorer prognosis, poorer AVT response, younger detection age, and death at a younger age compared to the patients with IPAH or HPAH carrying BMPR2 mutation.2) In the Japanese pediatric patients, HPAH carrying ALK-1 mutation was also reported to have a poor prognosis compared to IPAH.1)
According to these reports, it is clear that pediatric HPAH patients especially those carrying ALK-1 mutation have a severe disease with poor response to AVT and poor prognosis despite specific therapy for pulmonary arterial hypertension. Our patient also exhibited the same clinical characteristic as severe PAH in childhood, with poor response to AVT both in childhood and adulthood. However, he improved without any effective intervention and with a fair prognosis.
A case of self-limiting pediatric HPAH with endoglin(ENG) mutation has been reported.5) The patient was initially diagnosed as severe IPAH during infancy. Nine years later, his pulmonary arterial pressure decreased to almost normal level and he was diagnosed as a case of HHT carrying an ENG mutation. PAH carrying ENG mutation is now classified under HPAH in the current PAH guideline.6) In HHT with PAH, some patients might improve during childhood.
The one of the possible mechanisms of decreased PVR and pulmonary arterial pressure in our case was development of the shunt of pulmonary arterio-venous fistula originated from HHT. Our case presented SpO2 depletion on exercise testing performed at the age of 28 years. But his SpO2 at rest was within normal even when he was 28 years old. If the pulmonary vascular resistance decreased from 14.4 Wood units to 7.9 Wood units, large amount of intrapulmonary shunts should open and desaturation at rest would be observed.
ALK-1 is known as one of the components of BMPR2 -SMAD signal which influences proliferation of vascular endothelial cells and vascular smooth muscle cells. The mutation of ALK-1 is thought to mediate abnormal proliferation of pulmonary vascular endothelium and smooth muscle cells and cause pulmonary arterial hypertension. This pathway is known to be consist of many components and regulated from apelin, PPARγ, tenascin-C, adiponectin and other proteins.7) So this mutation may be influenced by environmental factor e.g., age. Among the patients with HHT which is also caused from the mutations of ALK-1/endoglin pathway, their frequent epistaxis usually begins in the age of 10 years and later increases in severity.8) This implies the ALK-1/endoglin pathway would be potentially influenced by the age. In addition to this, female patients with HHT1(caused by endoglin mutation) are reported to be severer and have shorter life.9) This also implies ALK-1/endoglin pathway is influenced by sex.
Our case fulfills the diagnostic criteria for HPAH and partially fulfills the criteria for HHT. Many HHT patients show no clinical signs of HHT in early childhood and on an average, the first symptoms are seen in the second decade of life.10) Hence, it is difficult to diagnose children with HHT at a younger age. Moreover, pediatric patients with PAH are reported to have a preserved cardiac index compared to adults with PAH; hence, symptoms of heart failure are not common in these patients.11) Therefore, pediatric patients with HHT associated with PAH in early childhood, are not easily diagnosed. In fact, we could not recognize him as the patient of HHT when he was six years old because he did not show other typical HHT findings except frequent nasal bleeding (desaturation due to pulmonary arteriovenous fistula, telangiectasia of skin, familial history).
It is thought that there will be a certain number of cases where PAH naturally improves and HHT is diagnosed at the age when the symptoms of HHT are exhibited. Although our case was diagnosed as PAH in childhood due to the symptoms of syncope and fatigue, we might have missed the HHT because he had minimal symptoms in the adult age.
We did not treat him with recent pulmonary vaso-dilator because his general status was good and he had not come to our outpatient clinic regularly despite our offer of regular visits. We found he could not continue to take pulmonary vaso-dilator in this good condition. If he shows even a small sign of clinical worsening in regular evaluation, we will administrate the disease specific drugs.
We experienced a case of HPAH carrying ALK-1 mutation who exhibited severe PAH in childhood, which improved during adulthood, without effective therapy. Although usually HPAH carrying ALK-1 gene mutation is severe in nature, our case showed a relatively good clinical course without treatment.
Our case is thought to be a case of HHT associated with PAH. Among the patients with HHT in adulthood, there are some cases who were associated with PAH in childhood and were under-diagnosed.
謝辞Acknowledgments
We thank Editage for English editing.
Conflicts of Interest
The authors declare that there is no conflict of interest.
引用文献References
1) Chida A, Shintani M, Yagi H, et al: Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am J Cardiol 2012; 110: 586–593
2) Girerd B, Montani D, Coulet F, et al: Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med 2010; 181: 851–861
3) Rich S, Dantzker DR, Ayres SM, et al: Primary pulmonary hypertension: A national prospective study. Ann Intern Med 1987; 107: 216–223
4) Barst RJ, McGoon MD, Elliott CG, et al: Survival in childhood pulmonary arterial hypertension: Insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation 2012; 125: 113–122
5) Mache CJ, Gamillscheg A, Popper HH, et al: Early-life pulmonary arterial hypertension with subsequent development of diffuse pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia type 1. Thorax 2008; 63: 85–86
6) Galiè N, Hoeper MM, Humbert M, et al: ESC Committee for Practice Guidelines (CPG): Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009; 30: 2493–2537
7) Rabinovitch M: Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 2012; 122: 4306–4313
8) Cottin V, Khouarta C, Dupuis-Girod S, et al: Pulmonary vascular disorders in hereditary hemorrhagic telangiectasia, in Humbert M(ed) : Pulmonary Vascular Disorders. Prog Respir Res. Basel Karger, 2012, pp 262–275
9) de Gussem EM, Edwards CP, Hoseman AE, et al: Life expectancy of parents with hereditary haemorrhagic telangiectasia. Orphanet J Rare Dis 2016; 11: 46–53
10) McCormick AA, Grundwaldt LJ: Vascular anomalies, in Zitelli BJMD, McIntire SCMD, Nowalk AJMDP (eds): Zitelli and Davis’ Atlas of Pediatric Physical Diagnosis, 2018, pp 378–393
11) Barst RJ, Ertel SI, Beghetti M, et al: Pulmonary arterial hypertension: A comparison between children and adults. Eur Respir J 2011; 37: 665–677
 1,Osamu Yamada1,Hideo Ohuchi1,Isao Shiraishi1,Hiroko Morisaki2,Takayuki Morisaki3,Ken-ichi Kurosaki1Toru Iwasa
1,Osamu Yamada1,Hideo Ohuchi1,Isao Shiraishi1,Hiroko Morisaki2,Takayuki Morisaki3,Ken-ichi Kurosaki1Toru Iwasa