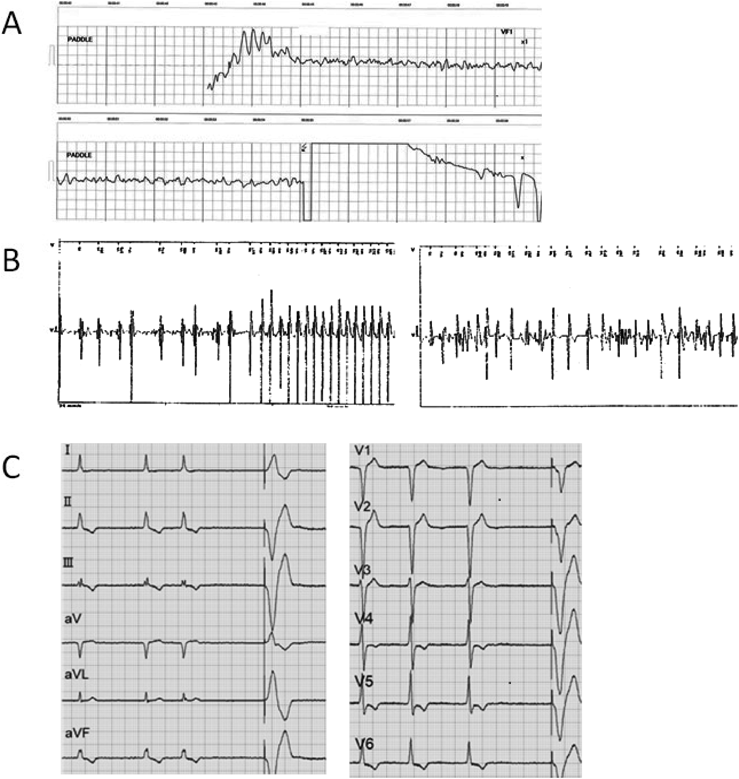
Ventricular Fibrillation in a Family with Short QT Syndrome Type 2 Carrying a Heterozygous KCNQ1-V141M Variant
1 Department of Pediatrics, Chiba Cerebral and Cardiovascular Center ◇ Ichihara, Japan
2 Department of Cardiovascular Medicine, Shiga University of Medical Science ◇ Otsu, Japan
3 Department of Community Medicine Supporting System, Kyoto University Graduate School of Medicine ◇ Kyoto, Japan
4 Department of Bioscience and Genetics, Cerebral and Cardiovascular Center ◇ Suita, Japan
5 Department of Cardiovascular Surgery, Chiba Cerebral and Cardiovascular Center ◇ Ichihara, Japan
6 Department of Cardiovascular Surgery, Chiba, Kaihin Municipal Hospital ◇ Chiba, Japan
7 Department of Pediatrics, Chiba Kaihin Municipal Hospital ◇ Chiba, Japan
受付日:2021年7月1日Received: July 1, 2021
受理日:2021年9月10日Accepted: September 10, 2021
発行日:2022年1月31日Published: January 31, 2022