Congenital heart defects (CHD) occur in nearly 1% of all live births and are the major cause of infant mortality and morbidity.1) Additionally, approximately 3 per 1,000 live births will require some intervention during the first year of life. Although the recent progress of comprehensive genetic studies could provide us a subset of etiologies of CHDs, the underlying pathogenesis of many CHDs has not been fully uncovered. The formation of the heart proceeds by sequential gene regulatory steps that dictate cell fates and organize specialized cell types into complex 3-dimensional units of structure and function. In order to explore the pathology of CHD, an approach focusing on the individual modular steps in cardiovascular morphogenesis is important, since most CHDs result from defective malformation in specific structural components of the developing heart and vessels. Series of studies about the disease modeling using transgenic animals provided us the insights into the cardiogenesis via regulation of essential cardiac transcription factors. Furthermore, human genetic studies revealed that the mutations of these cardiac transcription factors are frequently found in patients with familial CHDs. Among the cardiac transcription factors, members of the GATA family of zinc finger transcription factors and T-box transcription factors are thought as the core regulatory molecules for cardiogenesis and, to date, these factors are common genetic causes of various CHDs.
Developmental Origins of the Heart
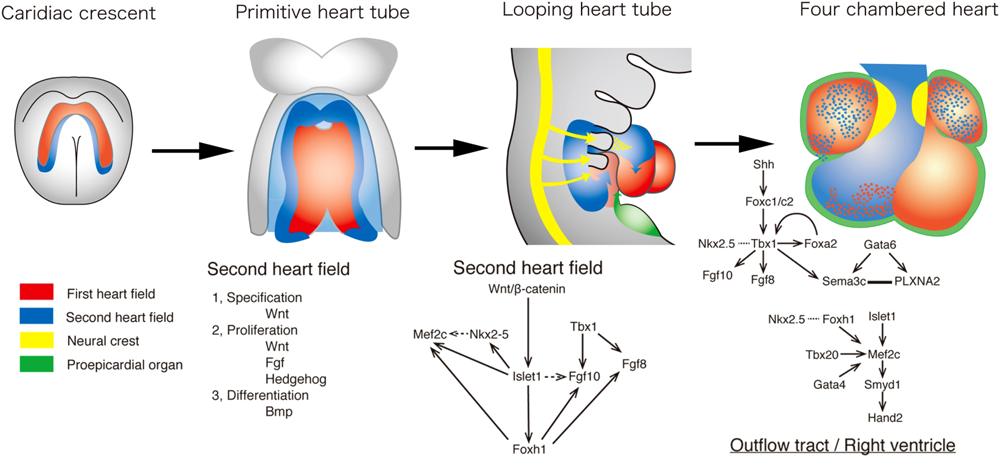
Current concept for developmental origins of the heart is shown in Fig. 1. Cells derived from the anterior lateral plate mesoderm form a crescent shape, called as the “first heart field (FHF),” at approximately embryonic (E) day 7.5 in the mouse embryo, corresponding to week 2 of human gestation.2, 3) By E8.0 in mice, or 3 weeks in humans, the FHF cells form a primitive heart tube, consisting of an interior layer of endocardial cells, an exterior layer of myocardial cells, and the cardiac jelly, the extracellular matrix for reciprocal signaling between the two layers. The FHF eventually contributes exclusively to the left ventricle and a part of atria.
In addition to the FHF, the second source of cardiac progenitor cells, termed the “second heart field (SHF)” lies medially to the cardiac crescent.2–4) During the primitive heart tube formation, the heart tube derived from the FHF may provide a scaffold upon which cells from the SHF migrate into both the arterial and venous poles of the heart tube. Upon rightward looping of the heart tube, SHF cells cross the pharyngeal mesoderm into the anterior and posterior portions, populating a large portion of the outflow tract (OFT) of the heart, the future right ventricle, and a part of atria.
Cardiac neural crest cells (cNCCs) originate from the dorsal neural tube between the mid-otic placode and the caudal boundary of the third somite.5) After they delaminate from the dorsal neural tube, cNCCs migrate into the caudal pharyngeal arches, and the OFT where they aggregate and form the OFT septum to separate the truncus arteriosus into two great arteries. cNCC is also localized to pharyngeal arches 3, 4 and 6, which give rise to the future great vessels. Many signaling pathways are involved in the migration and the condensation of cNCC, including reciprocal signaling between cNCC and the SHF that are essential for the development of the OFT and the aortic arch.6)
The fourth lineage of cardiac precursor cells is derived from the proepicardium (PE).7) The PE develops from the coelomic mesothelium that overlays the liver bud. During PE growth and epicardial formation, some PE/epicardial cells undergo an epithelial–mesenchymal transformation and are moved into the subepicardial space, giving rise to the precursors of the coronary vessels and connective tissue cells.8)
The GATA family is one of the major groups of zinc finger superfamily of transcription factors and conserves two zinc fingers that are necessary for binding to the unique DNA sequence (A/T)GATA(A/G) and the interaction with other transcription factors.9) In the GATA family, GATA4/5/6 are known to contribute to the cardiac development. GATA4 is the most investigated member, which is the predominant transcript in cardiomyocytes at all stages.10) Mice lacking Gata4 show embryonic lethality by E10.5 due to abnormal ventral folding, failure of midline fusion of the heart primordia, and extensive endoderm defects.10, 11) Decreasing expression of Gata4 leads to abnormal cardiac development with atrioventricular septal defect (AVSD), double outlet right ventricle (DORV), and hypoplasia of the ventricular myocardium in a dose-dependent manner.12)
Null mutations in Gata5 caused hypoplastic hearts and partially penetrant bicuspid aortic valves (BAV) formation.13) Gata5 is essential for the endocardial differentiation in a mouse cell line derived from cardiac mesoderm.14) In fact, expression pattern of Gata5 becomes restricted to the endocardium during development.14) Endocardial cell-specific inactivation of Gata5 led to BAV like null mutants.13)
Gata6 null mice die at E5.5–7.5 due to defects in extraembryonic endoderm.15, 16) Other knockdown experiments showed that GATA6 is required for differentiation of the cardiac lineage during embryogenesis in Xenopus and zebrafish.17) These results suggest that GATA6 is required at the earliest stage of development among the GATA family of proteins.
To date, the implication of GATA6 and GATA4 in CHD, as well as their genetic interaction, has been suggested. Genetic disruption of Gata6 in mice results in early embryonic lethality due to defects of endodermal differentiation. Conditional inactivation of Gata6 specifically in cNCCs causes persistent truncus arteriosus (PTA), suggesting an essential role of Gata6 during OFT development.18) Mice that are compound heterozygous for Gata4 and Gata6 null alleles die in utero and exhibit a spectrum of CHD, including septal and OFT defects.19) It has also been shown that Gata6 and Gata4 regulate their expression each other during development and that Gata6 may function in concert with Gata4 to direct tissue-specific gene expression essential for formation of the mammalian heart.
Besides the interaction between GATA4 and GATA6, many of the combinatorial interactions between the GATA factors and other cardiac transcription factors have been defined. Numerous studies have shown that Gata4 and Nkx2.5 directly interact to regulate the expression of the ANF, α-cardiac actin and cardiac restricted ankyrin repeat protein (CARP).20–23) Serum response factor (SRF) can physically interact with Gata4/5/6 or Nkx2.5 to synergistically activate the ANF and/or α-cardiac actin genes in cardiomyocytes.24–27) Gata4 recruited a subfamily of the MADS-box, myocyte enhancer factor-2 (MEF2) proteins to the ANF, α-cardiac actin, α-myosin heavy chain and BNP promoter and interact to activate the promoter synergistically.28) Gata4 also co-operate the ANF promoter with one of the cardiac essential T-box factor, Tbx5 similar to Nkx2.5,29) suggesting that a complex consisting of Tbx5, Nkx2.5 and Gata4 may function in the regulation of a subset of cardiac genes. In the mouse, Gata5 is the predominant factor expressed with Tbx20 in the endocardium and it has been shown to regulate endocardial differentiation in vitro,14) suggesting that the regulatory relationship between Tbx20 and Gata5 may be important in endocardial development.30) In summary, combinatorial interactions with other transcription factors involved in cardiogenesis are regulated by spatio-temporal and gene specific manner. The cardiac GATA factors orchestrate the synergistic regulatory cascade as central core factors.
Garg et al. first reported GATA4 mutations in patients with CHD and they found disturbance of interaction of GATA4 mutant proteins with TBX5.29) This study implicates a cooperative role for GATA4 and TBX5 in cardiac septation. Further linkage analyses were used to demonstrate associations between GATA4 mutations and multiple cardiac defects including atrial septal defect (ASD) and ventricular septal defect (VSD). As for GATA5, several genetic analyses revealed the association of GATA5 mutation with spectrum of CHDs similar to GATA4 mutation.31, 32)
Recently, we screened mutations of cardiac transcription factors in non-syndromic patients with PTA, and identified 2 different GATA6 mutations in 2 probands, but not in 182 unrelated controls with no CHD.33) Our subsequent biological analyses revealed that the expression of a neurovascular guiding molecule, SEMA3C, and its receptor, PLXNA2, were directly regulated by GATA6, and that both GATA6 mutant proteins failed to transactivate these genes. Transgenic analysis further suggested that the expression of SEMA3C and PLXNA2 in the OFT was dependent on GATA transcription factors during heart development. Together, our data implicate mutations in GATA6 as novel genetic causes of CHD involving the OFT, as a result of disruption of the direct regulation of semaphorin–plexin signaling (Fig. 1). After our study, there have been series of reports on mutations in GATA6 associated with CHD. Maitra et al. identified 2 novel sequence variations in GATA6 (A178V and L198V) in 2 individuals with tetralogy of Fallot (TOF) or AVSD from the screening of 310 individuals with non-syndromic CHD.34) Biochemical analysis demonstrated that the GATA6 A178V mutant protein resulted in increased transactivation ability for cardiac genes compared with the wild-type. Lin et al. identified a missense mutation (S184N) in GATA6 in 2 individuals with TOF or ASD.35) Allen et al. reported GATA6 mutations in patients with CHD including VSD, ASD or TOF complicated with pancreatic agenesis as well as congenital biliary tract anomalies, gut developmental disorders, and additional endocrine abnormalities.36) In mouse model, Gata6 and Gata4 contribute to the development of endoderm/mesoderm specification and differentiation.11, 37) Recent genetic analysis identified multiple GATA6 mutations in patients with CHD including TOF with pancreatic anomaly.38, 39) These findings implicate GATA6 in the development of multiple organ systems, including heart, especially the OFT and intracardiac septum, the biliary tract, gut, pituitary and thyroid, as well as the pancreas. Mutations of GATA genes associated with human CHD are summarized in Table 1.
Table 1 Mutations reported in core cardiac transcription factors in patients with non-syndromic CHD| Gene | Cardiac phenotype | Genotype | Ref. |
|---|
| GATA4 | ASD | P36S, H190R, S262A, V399G | 75) |
| PTA, PAVSD | T330R, S339R | 76) |
| VSD, TOF | G93A, Q316E, A411V, D425N | 77) |
| VSD | S335* | 78) |
| ASD | S52F, E359fs | 79) |
| ASD | C1074del | 80) |
| ASD, VSD, PS, AVSD, PDA, MR | E359del, G295S | 29) |
| ASD, VSD, PS | G296S | 81) |
| VSD, ASD, AVSD | F208L, F211L, G214S, M223T, P226fs, R229S, G234S, N239D, N239S, Y244C, N248S, R252P, I255T, R260Q, L261P, R260Q, L261P, Arg266*, N273S, T277I, R283H, N285K, C292R, A294V, H302R | 82) |
| TOF | E216D | 83) |
| VSD, TOF, AVSD | A6V, P163S, E359K, P407Q, S429T, A442V | 84) |
| ASD | c.341_342insA | 85) |
| ASD, VSD | H28Y, H436Y | 86) |
| ASD | T280M | 87) |
| ASD, VSD | G69D, P163R | 88) |
| ASD | G21V | 89) |
| VSD | G296R | 90) |
| VSD | R43W | 91) |
| VSD | Q55R, G96R, N197S, K404R | 92) |
| PS, TOF, VSD | A74D, G150W, E360G | 93) |
| ASD, PS | K319E | 94) |
| TOF | A9P, L51V, N285S | 95) |
| TOF, VSD | P163S, P407Q | 96) |
| ASD | M310V | 97) |
| GATA5 | TOF, ASD, PDA | R187G, H207R | 98) |
| BAV | Y16D, T252P | 99) |
| ASD, VSD, TOF, AS, DORV | R132G, V190A, N223H, H274R | 31) |
| VSD | L199V | 32) |
| GATA6 | TOF, ASD | S184N | 35) |
| ASD,VSD,PS,SVPS | Y323fs | 100) |
| PTA, ASD,PS,PDA | E486fs, N466H | 33) |
| PAVSD, MAPCA, DORV, VSD, PS, Dextrocardia | E142K | 76) |
| VSD | G220S | 101) |
| TOF, AVSD | A178V, L198V | 34) |
| VSD, TOF | D404Y, E460* | 102) |
| ASD, PDA | Y323fs, c.1428+1 G>T | 103) |
| TBX1 | DORV VSD | Q277* | 104) |
| PAVSD, PDA | E129K | 105) |
| PAVSD | c.1399-1428dup30 | 106) |
| PAVSD, IAA, VSD, ASD, MAPCA | F148Y, G310S, C1223del | 107) |
| TBX5 | TOF | S372L | 108) |
| TBX20 | ASD, aortic and mitoral valve defects | I121M | 68) |
| ASD, DCM, MVP, VSD | Q195*, I152M | 69) |
| ASD | D176N | 109) |
| DORV | R143W | 110) |
| ASD, TAPVC, TOF | A63T, I121F, T262M | 111) |
| ASD | Y309D, T370O, M395R | 112) |
| LVNC, DCM | Y317*, T262M | 61) |
T-box factors belong to an evolutionary conserved family of transcription factors which function as repressors or activators. The spatiotemporal expression of T-box genes in the cardiogenic region modulates key steps for genesis of the cardiac components. These factors play an essential role in the development of cardiac progenitor cells as well as in the patterning of chambers.40) Tbx1/2/3/5/18/20 are the major factors that are associated with the heart development in mammals, birds, fish, and amphibians.40)
The expression program in the SHF appears to be modulated by T-box factors. Recent progress of transgenic mouse model revealed that a T-box transcription factor, Tbx1, is a major genetic determinant of cardiac and craniofacial disorders associated with Tbx1 is expressed in the SHF, but not in the FHF or cNCC.46) OFT defects, typically PTA and TOF, and aortic arch anomalies are highly associated with 22q11DS, as well as craniofacial defects including cleft palate. Tbx1 null mice phenocopy the 22q11DS phenotype. Tissue-specific disruption of Tbx1 in the expression domain of Nkx2.5 developed the single OFT with no evidence of an aortopulmonary septum, suggesting that loss of Tbx1 function in the SHF or pharyngeal endoderm may result in OFT defects.47) The expression of Tbx1 in the SHF is directly regulated by forkhead-containing (Fox) transcription factors, Foxa2, Foxc1 and Foxc248, 49) (Fig. 1). Shh plays a role in maintenance of the expression of Tbx1 in the upstream of Fox factors.46, 48) The Tbx1 hypomorphic mouse demonstrated that the development of the OFT was sensitive to Tbx1 dosage.50) In experiments using knockout mice, Foxc1 and Foxc2, and Foxh1 were involved in the development of the OFT, although Foxa2-null mice die too early in utero to study cardiac development. Foxc1 or Foxc2 null mice and compound heterozygous mice for Foxc1 and Foxc2 display aortic arch defects reminiscent of Tbx1 heterozygous mice.51) Like Tbx1 hypomorphic mice, they also display a reduction in the size of the OFT, suggesting a dose-dependent regulatory mechanism of Foxc1 and Foxc2 in the SHF.52) Moreover, embryos homozygous-null for both Foxc1 and Foxc2 display an absence of the OFT and right ventricle associated with downregulation of Tbx1 and its downstream effectors, Fgf8 and 10 (Fig. 1).
Tbx18 is expressed in progenitors that contribute to the PE and the mesenchyme that borders the inflow tract, specifically the sinus venosus.40, 53, 54) Coronary smooth muscles and cardiac fibroblasts are also derived from Tbx18 positive progenitors.55)
Tbx20 is expressed in a subset of SHF precursors and mice deficient for Tbx20 show outflow tract defects with short heart tubes, suggesting failure in the development of SHF progenitors.40, 56, 57) A protein–protein interaction between Tbx20 and Gata4 induces Nkx2.5 and Mef2c in the right ventricle and outflow tract, whereas these factors were severely downregulated in Tbx20 null embryos58) with an ectopic upregulation of Tbx2.59) Tbx2 represses the ventricular chamber specific markers via an interaction with Tbx and contributes to specify the non-chamber region including segmentation of atrioventricular boundary. These results suggest that Tbx20 contributes to the stable expression of chamber specific genes by inhibition of Tbx2. Tbx20 also plays a role for the proliferative expansion of the chambers via induction of Nmyc160) and thought to be a key factor for the development of embryonic myocardium.
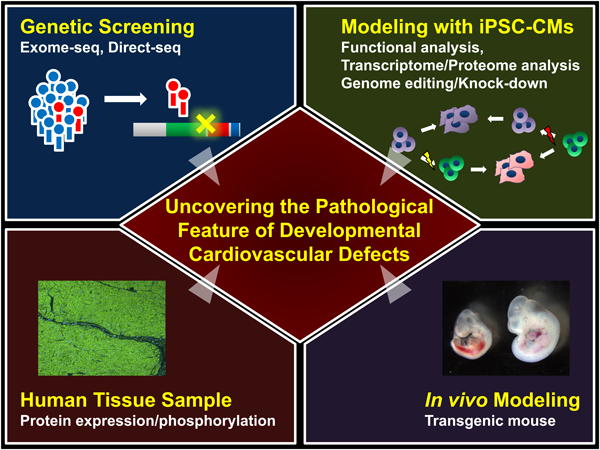
Recently, we performed genetic tests in left ventricular non-compaction (LVNC) cardiomyopathy patients and identified two independent TBX20 mutations from a family with LVNC and another isolated patient.61) LVNC has a unique structural phenotype including a characteristic deep and extensive hypertrabeculation of the LV. LVNC has been theorized to result from the arrest of compaction of the developing LV myocardium, as it passes through several distinct evolutionally conserved steps, and thought to have a potential as the good model to understand the developmental mechanism of myocardium. We generated patient-specific induced pluripotent stem cells (iPSCs) from the family members and induced cardiac differentiation. LVNC patient-specific iPSC-derived cardiomyocytes have decreased cell proliferation which prevents proper development of the embryonic heart and which is associated with abnormal upregulation of TGFβ signaling. Abnormal activation of TGFβ signaling in embryonic myocardium causes growth arrest of the developing heart in vivo. Functional disturbance of TBX20 prevents proper regulation of TGFβ signaling and contributes to the LVNC phenotype. Importantly, we showed that LVNC iPSCs could be rescued using drugs targeting TGFβ signaling or by genetic modification of the TBX20 mutation. These results clearly show the essential role of TBX20 in embryonic myocardium growth and ventricular chamber development. This study suggests that iPSC technology is a powerful tool for uncovering the pathological mechanisms of CHD (Fig. 2).
Tbx5 is an essential factor for the morphological development of the heart as well as the development of the cardiac conduction system.62) Like the other core cardiac transcription factors, Tbx5 is involved in multiple transcription factor pathways and combinatorial interactions with other cardiac transcription factors. Tbx5 knockout mouse embryos have abnormal heart tube formation with hypoplastic atria.63) Mice with haploinsufficiency of Tbx5 have ASD with or without VSD, and atrioventricular (AV).63) Mice doubly heterozygous for Tbx5 and Gata4 have growth retardation and early neonatal lethality with AVSD and myocardial thinning, similar to the human GATA4 mutant phenotype.64) Interactions between Tbx5/20, Gata4 and Nkx2.5 have an important role in the activation of chamber specification and upregulation of the chamber-specific markers, including Nppa.65, 66)
Regarding the CHD in human, TBX1 mutations are associated with the cardiac phenotype of DiGeorge syndrome (DGS), including outflow tract and conotruncal defects as well as great vessel mis-patterning.41, 42) Mutations in TBX5 are linked to the Holt–Oram syndrome (HOS) characterized by forelimb and heart defects. The aberrant cardiac phenotype manifests in atrial and ventricular septal defectsas well as conduction system disturbance.67) TBX20 mutations correlate with aberrant valvulogenesis and septal defects.68, 69) These mutations, however, can lead to either a gain or a loss of function, suggesting an important role of gene and protein dosage in regulating different aspects of chamber formation and subsequent function of the heart.68) Interestingly, TBX3 mutations are correlated with the ulnar-mammary syndrome (UMS) with rare evident cardiac defects only two case studies identified UMS patients with overt ventricular septal defects (VSD) as well as cardiac conduction disturbance70, 71). This may be due to a functional redundancy with TBX2. To date, there is no report about the mutation in TBX2 gene, however, recent two studies showed that duplication or deletion of genomic region including TBX2 gene may be associated with CHD in human.72, 73) TBX3 may also be associated with the left ventricular mass in human.74) Mutations of T-box genes associated with human CHD are summarized in Table 1.
Through the use of animal models combined with human genetic investigations, a molecular framework via core transcription factors has gradually uncovered the detailed steps of cardiovascular morphogenesis. Such advances lead to an approach to the etiology of “multifactorial” CHD in the post-genomic era. However, it is still challenging to use the resources derived from cardiac tissues or cells of patients with CHD and reveal molecular mechanisms underlying the pathogenesis of 3D-structural CHD in human. Recent progress of comprehensive analysis using deep-sequencing would provide more detailed network between core cardiac transcription factors and their effectors that influence the phenotype, and eventually lead to the preventive and/or regenerative medicine for CHD.
謝辞Acknowledgments
We thank D. Srivastava, R. Matsuoka, D. Bernstein and J.C. Wu for their support with the projects in this review. This work is supported by the Uehara Memorial Foundation Research Fellowship, American Heart Association Postdoctoral Fellowship, the Encouraging Development of Strategic Research Centers, MEXT KAKENHI, Grant-in-Aid for Scientific Research (B) and Grant-in-Aid for Young Scientists (B), and Special Coordination Funds for Promoting Science and Technology, Ministry of Education, Culture, Sports, Science, and Technology.
Conflicts of Interest
The authors have no conflicts of interest to declare.
引用文献References
1) Hoffman JI, Kaplan S: The incidence of congenital heart disease. J Am Coll Cardiol 2002; 39: 1890–1900
2) Srivastava D: Making or breaking the heart: From lineage determination to morphogenesis. Cell 2006; 126: 1037–1048
3) Buckingham M, Meilhac S, Zaffran S: Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet 2005; 6: 826–835
4) Kelly RG, Brown NA, Buckingham ME: The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev Cell 2001; 1: 435–440
5) Hutson MR, Kirby ML: Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin Cell Dev Biol 2007; 18: 101–110
6) Waldo KL, Hutson MR, Stadt HA, et al: Cardiac neural crest is necessary for normal addition of the myocardium to the arterial pole from the secondary heart field. Dev Biol 2005; 281: 66–77
7) Mikawa T, Gourdie RG: Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev Biol 1996; 174: 221–232
8) Olivey HE, Svensson EC: Epicardial-myocardial signaling directing coronary vasculogenesis. Circ Res 2010; 106: 818–832
9) Molkentin JD: The zinc finger-containing transcription factors GATA-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem 2000; 275: 38949–38952
10) Molkentin JD, Lin Q, Duncan SA, et al: Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev 1997; 11: 1061–1072
11) Kuo CT, Morrisey EE, Anandappa R, et al: GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev 1997; 11: 1048–1060
12) Pu WT, Ishiwata T, Juraszek AL, et al: GATA4 is a dosage-sensitive regulator of cardiac morphogenesis. Dev Biol 2004; 275: 235–244
13) Laforest B, Andelfinger G, Nemer M: Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin Invest 2011; 121: 2876–2887
14) Nemer G, Nemer M: Cooperative interaction between GATA5 and NF-ATc regulates endothelial-endocardial differentiation of cardiogenic cells. Development 2002; 129: 4045–4055
15) Morrisey EE, Tang Z, Sigrist K, et al: GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes Dev 1998; 12: 3579–3590
16) Koutsourakis M, Langeveld A, Patient R, et al: The transcription factor GATA6 is essential for early extraembryonic development. Development 1999; 126: 723–732
17) Peterkin T, Gibson A, Patient R: GATA-6 maintains BMP-4 and Nkx2 expression during cardiomyocyte precursor maturation. EMBO J 2003; 22: 4260–4273
18) Lepore JJ, Mericko PA, Cheng L, et al: GATA-6 regulates semaphorin 3C and is required in cardiac neural crest for cardiovascular morphogenesis. J Clin Invest 2006; 116: 929–939
19) Xin M, Davis CA, Molkentin JD, et al: A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proc Natl Acad Sci USA 2006; 103: 11189–11194
20) Durocher D, Nemer M: Combinatorial interactions regulating cardiac transcription. Dev Genet 1998; 22: 250–262
21) Durocher D, Charron F, Warren R, et al: The cardiac transcription factors Nkx2-5 and GATA-4 are mutual cofactors. EMBO J 1997; 16: 5687–5696
22) Sepulveda JL, Belaguli N, Nigam V, et al: GATA-4 and Nkx-2.5 coactivate Nkx-2 DNA binding targets: Role for regulating early cardiac gene expression. Mol Cell Biol 1998; 18: 3405–3415
23) Kuo H, Chen J, Ruiz-Lozano P, et al: Control of segmental expression of the cardiac-restricted ankyrin repeat protein gene by distinct regulatory pathways in murine cardiogenesis. Development 1999; 126: 4223–4234
24) Belaguli NS, Sepulveda JL, Nigam V, et al: Cardiac tissue enriched factors serum response factor and GATA-4 are mutual coregulators. Mol Cell Biol 2000; 20: 7550–7558
25) Morin S, Paradis P, Aries A, et al: Serum response factor-GATA ternary complex required for nuclear signaling by a G-protein-coupled receptor. Mol Cell Biol 2001; 21: 1036–1044
26) Sepulveda JL, Vlahopoulos S, Iyer D, et al: Combinatorial expression of GATA4, Nkx2-5, and serum response factor directs early cardiac gene activity. J Biol Chem 2002; 277: 25775–25782
27) Nishida W, Nakamura M, Mori S, et al: A triad of serum response factor and the GATA and NK families governs the transcription of smooth and cardiac muscle genes. J Biol Chem 2002; 277: 7308–7317
28) Morin S, Charron F, Robitaille L, et al: GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J 2000; 19: 2046–2055
29) Garg V, Kathiriya IS, Barnes R, et al: GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003; 424: 443–447
30) Stennard FA, Costa MW, Elliott DA, et al: Cardiac T-box factor Tbx20 directly interacts with Nkx2-5, GATA4, and GATA5 in regulation of gene expression in the developing heart. Dev Biol 2003; 262: 206–224
31) Jiang JQ, Li RG, Wang J, et al: Prevalence and spectrum of GATA5 mutations associated with congenital heart disease. Int J Cardiol 2013; 165: 570–573
32) Wei D, Bao H, Zhou N, et al: GATA5 loss-of-function mutation responsible for the congenital ventriculoseptal defect. Pediatr Cardiol 2013; 34: 504–511
33) Kodo K, Nishizawa T, Furutani M, et al: GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc Natl Acad Sci USA 2009; 106: 13933–13938
34) Maitra M, Koenig SN, Srivastava D, et al: Identification of GATA6 sequence variants in patients with congenital heart defects. Pediatr Res 2010; 68: 281–285
35) Lin X, Huo Z, Liu X, et al: A novel GATA6 mutation in patients with tetralogy of Fallot or atrial septal defect. J Hum Genet 2010; 55: 662–667
36) Lango Allen H, Flanagan SE, Shaw-Smith C, et al; International Pancreatic Agenesis Consortium: GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet 2011; 44: 20–22
37) Xuan S, Sussel L: GATA4 and GATA6 regulate pancreatic endoderm identity through inhibition of hedgehog signaling. Development 2016; 143: 780–786
38) Yau D, De Franco E, Flanagan SE, et al: Case report: maternal mosaicism resulting in inheritance of a novel GATA6 mutation causing pancreatic agenesis and neonatal diabetes mellitus. Diagn Pathol 2017; 12: 1
39) Gong M, Simaite D, Kuhnen P, et al: Two novel GATA6 mutations cause childhood-onset diabetes mellitus, pancreas malformation and congenital heart disease. Horm Res Paediatr 2013; 79: 250–256
40) Hoogaars WM, Barnett P, Moorman AF, et al: T-box factors determine cardiac design. Cell Mol Life Sci 2007; 64: 646–660
41) Yamagishi H, Srivastava D: Unraveling the genetic and developmental mysteries of 22q11 deletion syndrome. Trends Mol Med 2003; 9: 383–389
42) Yamagishi H: The 22q11.2 deletion syndrome. Keio J Med 2002; 51: 77–88
43) Jerome LA, Papaioannou VE: DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet 2001; 27: 286–291
44) Lindsay EA, Vitelli F, Su H, et al: Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001; 410: 97–101
45) Merscher S, Funke B, Epstein JA, et al: TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 2001; 104: 619–629
46) Garg V, Yamagishi C, Hu T, et al: Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev Biol 2001; 235: 62–73
47) Xu H, Morishima M, Wylie JN, et al: Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development 2004; 131: 3217–3227
48) Yamagishi H, Maeda J, Hu T, et al: Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev 2003; 17: 269–281
49) Maeda J, Yamagishi H, McAnally J, et al: Tbx1 is regulated by forkhead proteins in the secondary heart field. Dev Dyn 2006; 235: 701–710
50) Hu T, Yamagishi H, Maeda J, et al: Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development 2004; 131: 5491–5502
51) Kume T, Jiang H, Topczewska JM, et al: The murine winged helix transcription factors, Foxc1 and Foxc2, are both required for cardiovascular development and somitogenesis. Genes Dev 2001; 15: 2470–2482
52) Seo S, Kume T: Forkhead transcription factors, Foxc1 and Foxc2, are required for the morphogenesis of the cardiac outflow tract. Dev Biol 2006; 296: 421–436
53) Kraus F, Haenig B, Kispert A: Cloning and expression analysis of the mouse T-box gene Tbx18. Mech Dev 2001; 100: 83–86
54) Mommersteeg MT, Dominguez JN, Wiese C, et al: The sinus venosus progenitors separate and diversify from the first and second heart fields early in development. Cardiovasc Res 2010; 87: 92–101
55) Cai CL, Martin JC, Sun Y, et al: A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008; 454: 104–108
56) Iio A, Koide M, Hidaka K, et al: Expression pattern of novel chick T-box gene, Tbx20. Dev Genes Evol 2001; 211: 559–562
57) Chen JN, van Eeden FJ, Warren KS, et al: Left-right pattern of cardiac BMP4 may drive asymmetry of the heart in zebrafish. Development 1997; 124: 4373–4382
58) Takeuchi JK, Mileikovskaia M, Koshiba-Takeuchi K, et al: Tbx20 dose-dependently regulates transcription factor networks required for mouse heart and motoneuron development. Development 2005; 132: 2463–2474
59) Stennard FA, Costa MW, Lai D, et al: Murine T-box transcription factor Tbx20 acts as a repressor during heart development, and is essential for adult heart integrity, function and adaptation. Development 2005; 132: 2451–2462
60) Cai CL, Zhou W, Yang L, et al: T-box genes coordinate regional rates of proliferation and regional specification during cardiogenesis. Development 2005; 132: 2475–2487
61) Kodo K, Ong SG, Jahanbani F, et al: iPSC-derived cardiomyocytes reveal abnormal TGF-beta signalling in left ventricular non-compaction cardiomyopathy. Nat Cell Biol 2016; 18: 1031–1042
62) Hatcher CJ, Basson CT: Specification of the cardiac conduction system by transcription factors. Circ Res 2009; 105: 620–630
63) Bruneau BG, Nemer G, Schmitt JP, et al: A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 2001; 106: 709–721
64) Maitra M, Schluterman MK, Nichols HA, et al: Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev Biol 2009; 326: 368–377
65) Christoffels VM, Burch JB, Moorman AF: Architectural plan for the heart: Early patterning and delineation of the chambers and the nodes. Trends Cardiovasc Med 2004; 14: 301–307
66) Naiche LA, Harrelson Z, Kelly RG, et al: T-box genes in vertebrate development. Annu Rev Genet 2005; 39: 219–239
67) Mori AD, Bruneau BG: TBX5 mutations and congenital heart disease: Holt-Oram syndrome revealed. Curr Opin Cardiol 2004; 19: 211–215
68) Posch MG, Gramlich M, Sunde M, et al: A gain-of-function TBX20 mutation causes congenital atrial septal defects, patent foramen ovale and cardiac valve defects. J Med Genet 2010; 47: 230–235
69) Kirk EP, Sunde M, Costa MW, et al: Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet 2007; 81: 280–291
70) Linden H, Williams R, King J, et al: Ulnar Mammary syndrome and TBX3: Expanding the phenotype. Am J Med Genet A 2009; 149A: 2809–2812
71) Meneghini V, Odent S, Platonova N, et al: Novel TBX3 mutation data in families with ulnar-mammary syndrome indicate a genotype-phenotype relationship: mutations that do not disrupt the T-domain are associated with less severe limb defects. Eur J Med Genet 2006; 49: 151–158
72) Radio FC, Bernardini L, Loddo S, et al: TBX2 gene duplication associated with complex heart defect and skeletal malformations. Am J Med Genet A 2010; 152A: 2061–2066
73) Ballif BC, Theisen A, Rosenfeld JA, et al: Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am J Hum Genet 2010; 86: 454–461
74) Sano M, Kamitsuji S, Kamatani N, et al; Japan Pharmacogenomics Data Science Consortium (JPDSC): Genome-wide association study of absolute QRS voltage identifies common variants of TBX3 as genetic determinants of left ventricular mass in a healthy Japanese Population. PLoS ONE 2016; 11: e0155550
75) Yang YQ, Wang J, Liu XY, et al: Mutation spectrum of GATA4 associated with congenital atrial septal defects. Arch Med Sci 2013; 9: 976–983
76) Kodo K, Nishizawa T, Furutani M, et al: Genetic analysis of essential cardiac transcription factors in 256 patients with non-syndromic congenital heart defects. Circ J 2012; 76: 1703–1711
77) Tomita-Mitchell A, Maslen CL, Morris CD, et al: GATA4 sequence variants in patients with congenital heart disease. J Med Genet 2007; 44: 779–783
78) Cheng C, Lin Y, Yang F, et al: Mutational screening of affected cardiac tissues and peripheral blood cells identified novel somatic mutations in GATA4 in patients with ventricular septal defect. J Biomed Res 2011; 25: 425–430
79) Hirayama-Yamada K, Kamisago M, Akimoto K, et al: Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet A 2005; 135: 47–52
80) Okubo A, Miyoshi O, Baba K, et al: A novel GATA4 mutation completely segregated with atrial septal defect in a large Japanese family. J Med Genet 2004; 41: e97
81) Sarkozy A, Conti E, Neri C, et al: Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet 2005; 42: e16
82) Reamon-Buettner SM, Borlak J: GATA4 zinc finger mutations as a molecular rationale for septation defects of the human heart. J Med Genet 2005; 42: e32
83) Nemer G, Fadlalah F, Usta J, et al: A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat 2006; 27: 293–294
84) Zhang W, Li X, Shen A, et al: GATA4 mutations in 486 Chinese patients with congenital heart disease. Eur J Med Genet 2008; 51: 527–535
85) Hamanoue H, Rahayuningsih SE, Hirahara Y, et al: Genetic screening of 104 patients with congenitally malformed hearts revealed a fresh mutation of GATA4 in those with atrial septal defects. Cardiol Young 2009; 19: 482–485
86) Chen MW, Pang YS, Guo Y, et al: GATA4 mutations in Chinese patients with congenital cardiac septal defects. Pediatr Cardiol 2010; 31: 85–89
87) Chen Y, Mao J, Sun Y, et al: A novel mutation of GATA4 in a familial atrial septal defect. Clin Chim Acta 2010; 411: 1741–1745
88) Butler TL, Esposito G, Blue GM, et al: GATA4 mutations in 357 unrelated patients with congenital heart malformation. Genet Test Mol Biomarkers 2010; 14: 797–802
89) Liu XY, Wang J, Zheng JH, et al: Involvement of a novel GATA4 mutation in atrial septal defects. Int J Mol Med 2011; 28: 17–23
90) Wang J, Fang M, Liu XY, et al: A novel GATA4 mutation responsible for congenital ventricular septal defects. Int J Mol Med 2011; 28: 557–564
91) Yang YQ, Li L, Wang J, et al: A novel GATA4 loss-of-function mutation associated with congenital ventricular septal defect. Pediatr Cardiol 2012; 33: 539–546
92) Yang YQ, Wang J, Liu XY, et al: Novel GATA4 mutations in patients with congenital ventricular septal defects. Med Sci Monit 2012; 18: CR344–CR350
93) Wang E, Sun S, Qiao B, et al: Identification of functional mutations in GATA4 in patients with congenital heart disease. PLoS ONE 2013; 8: e62138
94) Xiang R, Fan LL, Huang H, et al: A novel mutation of GATA4 (K319E) is responsible for familial atrial septal defect and pulmonary valve stenosis. Gene 2013
95) Yang YQ, Gharibeh L, Li RG, et al: GATA4 loss-of-function mutations underlie familial tetralogy of fallot. Hum Mutat 2013; 34: 1662–1671
96) Peng T, Wang L, Zhou SF, et al: Mutations of the GATA4 and NKX2.5 genes in Chinese pediatric patients with non-familial congenital heart disease. Genetica 2010; 138: 1231–1240
97) Chen Y, Han ZQ, Yan WD, et al: A novel mutation in GATA4 gene associated with dominant inherited familial atrial septal defect. J Thorac Cardiovasc Surg 2010; 140: 684–687
98) Wei D, Bao H, Liu XY, et al: GATA5 loss-of-function mutations underlie tetralogy of fallot. Int J Med Sci 2013; 10: 34–42
99) Shi LM, Tao JW, Qiu XB, et al: GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int J Mol Med 2014; 33: 1219–1226
100) Eifes S, Chudasama KK, Molnes J, et al: A novel GATA6 mutation in a child with congenital heart malformation and neonatal diabetes. Clin Case Rep 2013; 1: 86–90
101) Zheng GF, Wei D, Zhao H, et al: A novel GATA6 mutation associated with congenital ventricular septal defect. Int J Mol Med 2012; 29: 1065–1071
102) Wang J, Luo XJ, Xin YF, et al: Novel GATA6 mutations associated with congenital ventricular septal defect or tetralogy of fallot. DNA Cell Biol 2012; 31: 1610–1617
103) Chao CS, McKnight KD, Cox KL, et al: Novel GATA6 mutations in patients with pancreatic agenesis and congenital heart malformations. PLoS ONE 2015; 10: e0118449
104) Pan Y, Wang ZG, Liu XY, et al: A novel TBX1 loss-of-function mutation associated with congenital heart disease. Pediatr Cardiol 2015; 36: 1400–1410
105) Xu YJ, Chen S, Zhang J, et al: Novel TBX1 loss-of-function mutation causes isolated conotruncal heart defects in Chinese patients without 22q11.2 deletion. BMC Med Genet 2014; 15: 78
106) Rauch R, Hofbeck M, Zweier C, et al: Comprehensive genotype–phenotype analysis in 230 patients with tetralogy of Fallot. J Med Genet 2010; 47: 321–331
107) Yagi H, Furutani Y, Hamada H, et al: Role of TBX1 in human del22q11.2 syndrome. Lancet 2003; 362: 1366–1373
108) Baban A, Postma AV, Marini M, et al: Identification of TBX5 mutations in a series of 94 patients with Tetralogy of Fallot. Am J Med Genet A 2014; 164A: 3100–3107
109) Liu JJ, Fan LL, Chen JL, et al: A novel variant in TBX20 (p.D176N) identified by whole-exome sequencing in combination with a congenital heart disease related gene filter is associated with familial atrial septal defect. J Zhejiang Univ Sci B 2014; 15: 830–837
110) Pan Y, Geng R, Zhou N, et al: TBX20 loss-of-function mutation contributes to double outlet right ventricle. Int J Mol Med 2015; 35: 1058–1066
111) Liu C, Shen A, Li X, et al: T-box transcription factor TBX20 mutations in Chinese patients with congenital heart disease. Eur J Med Genet 2008; 51: 580–587
112) Monroy-Munoz IE, Perez-Hernandez N, Rodriguez-Perez JM, et al: Novel mutations in the transcriptional activator domain of the human TBX20 in patients with atrial septal defect. Biomed Res Int 2015; 2015: 718786