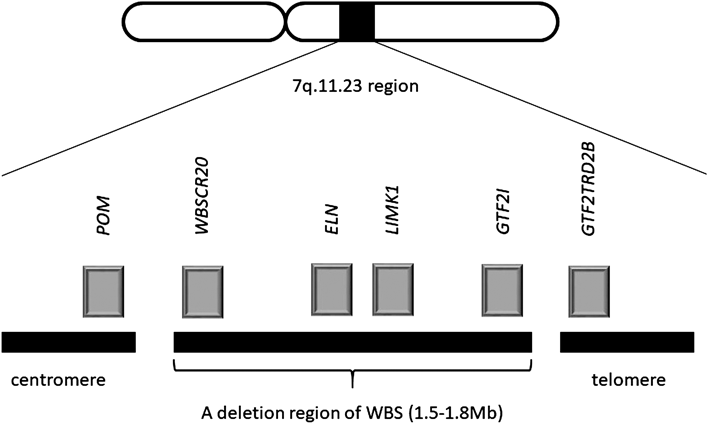
Williams-Beuren syndrome (WBS) is contiguous gene syndrome caused by microdeletions of 7q11.23 and features cardiovascular lesions (supravalvular aortic stenosis [SVAS] and peripheral pulmonary artery stenosis [PPS]), distinctive facies (thick medial eyebrow, blepharophimosis, epicanthus, puffy eye, short and turned-up nose, long philtrum, opened thick lips) and specific cognitive profiles (specialty of the expressed languages, recognition of faces and music and poor visuospatial perception). The frequency of WBS has been estimated to be 1: 7500–20000, no gender differences, and the majority of them is sporadic. The prognosis of WBS without cardiovascular lesions is relatively favorable.1) The chromosome 7q11.23 includes several genes, ELN and GTF2I (Fig. 1), and ELN is responsible for expressions of the elastin which is the main component of elastic fibers. The elastin is comprised of systemic connective tissue, aorta, lungs, skins and ligaments, and the abnormal expressions of elastin according to ELN mutation induce various symptoms, example for vascular wall thickening. The past reports of ELN failure are accompanied with WBS and SVAS (familial and sporadic).
Here, we present the case of a girl with a nonsense mutation in ELN that has never been reported before, while she has only characteristics cardiovascular lesions of WBS and fluorescence in situ hybridization (FISH) negative.
A baby girl was not pointed out fetal abnormality and had nonconsanguineous healthy parents, two sisters and one brother. She was born at 40 weeks of gestation by vaginal delivery at previous hospital. After birth, she was systemic cyanosis and crying weakly, so needed manual ventilation (Apgar score: 6–8). She was pointed out systolic murmur and transferred our hospital.
Her clinical findings was as follows; body temperature was 37.1°C, pulse rate was 104 beats per minute, percutaneous saturation was 99–100% (oxygen flow 1 L per minute), blood pressure was 92/50 mmHg, height was 48 cm (−0.91 SD), body weight was 3,374 g (+0.61 SD) and head circumference was 33 cm (−0.39 SD). Her large fontanel was 2 cm and flat. She did not have cyanosis and distinctive facies. Her respiratory sounds were good, and systolic murmur (Levine III/VI) at three left sternal bone were noticed. Her abdomen was soft and flat, and good bowel sounds without hepatosplenomegaly, tumor and abdominal vascular sounds. An edema, joint contracture and skin lesion was not noticed. Laboratory routine investigations in blood were normal. A cardiothoracic rate was 0.66 without pulmonary lesions by X-ray investigation. ECG showed the right axis deviation and R-wave progression in V3 induction. 2D echo showed a right ventricular hyperplasia, a tricuspid regurgitation and a pressure gradient between right atrium and ventricle. A diameter of supravalvular pulmonary artery was 3.2 mm with a pressure gradient between the stenosis. A bifurcation of pulmonary trunk stenosis made prominent mosaic blood flow in peripheral pulmonary arteries. An Aortic valve was tricuspid without regurgitation. A stenosis of sinotubular junction, 3.9 mm (−2.27 SD), 2.7 meter per second and estimated 30 mmHg pressure gradient, made a mosaic blood flow after the stenosis. In conclusion, we diagnosed her SVAS, PPS and supravalvular pulmonary stenosis.
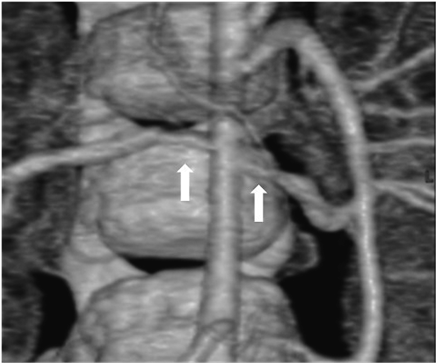
Her state was stable without particular interventions until echocardiography showed the signs of right heart failure at six days after birth, and we began diuretics (furosemide [1 mg/kg/day] and aldactone [1 mg/kg/day]). She was discharged at twenty days after birth since SVAS and PPS did not proceed. She was a normal psychomotor development, smiling at two months old, head controlling at four months old, sitting at seven months old and crawling at nine months old. The distinctive facies and the undeveloped recognition were not seen in follow-up periods. At ten months old, she was hospitalized and received cardiac catheterization for consideration of treatment policy. Cardiac catheterization revealed that the pressure gradient was right atrium-right ventricle, bilateral peripheral pulmonary arteries-main pulmonary artery and left ventricle-ascending aorta. Estimated pressure gradient between left ventricle and ascending aorta was 40 mmHg and cardiac contractility was sufficient (Table 1), so we decided to manage her without surgical interventions. For hypertension, we performed Multi Detector-row Computed Tomography and discovered bilateral renal artery stenosis (Fig. 2). She had high renin/angiotensin ratio (plasma renin activity was 59.5 ng/mL/hr [reference range; 0.2–2.7 ng/mL/hr] and plasma aldosterone concentration was 128 ng/dL [reference range; 2–13 ng/dL]), and we diagnosed her renovascular hypertension and started carvedilol.
Table 1 Cardiac catheterization at 10 months| Site | Pressure (mmHg) |
|---|
| RV | 78/4 |
| LV | 148/10 |
| aAo | 112/48 (76) |
| main PA | 74/10 (32) |
| rt. PA | 17/6 (8) |
| lt. PA | 16/10 (11) |
| Wedge mean | R=8 | L=8 |
| RV-MPA=70 mmHg |
| lt.PA-main PA=59 mmHg |
| rt.PA prox-main PA=40 mmHg |
| rt.PA prox-RPA dist=17 mmHg |
| aAo: ascending aorta, LV: left ventricle, PA: pulmonary artery, RV: right ventricle |
She is now one and a half years old, and height is 80.6 cm (+0.44 SD), body weight is 9.1 kg (−0.78 SD) and head circumference was 44.8 cm (−1.13 SD). She has good cardiac function without hypertension and is not psychomotor retardation (movement; 94, hand movement; 108, basic habit; 108, interpersonal relation; 108, speech; 94 and language understanding; 94 by using Enjoji Scale of Infant Analytical development).
Molecular Genetic Study
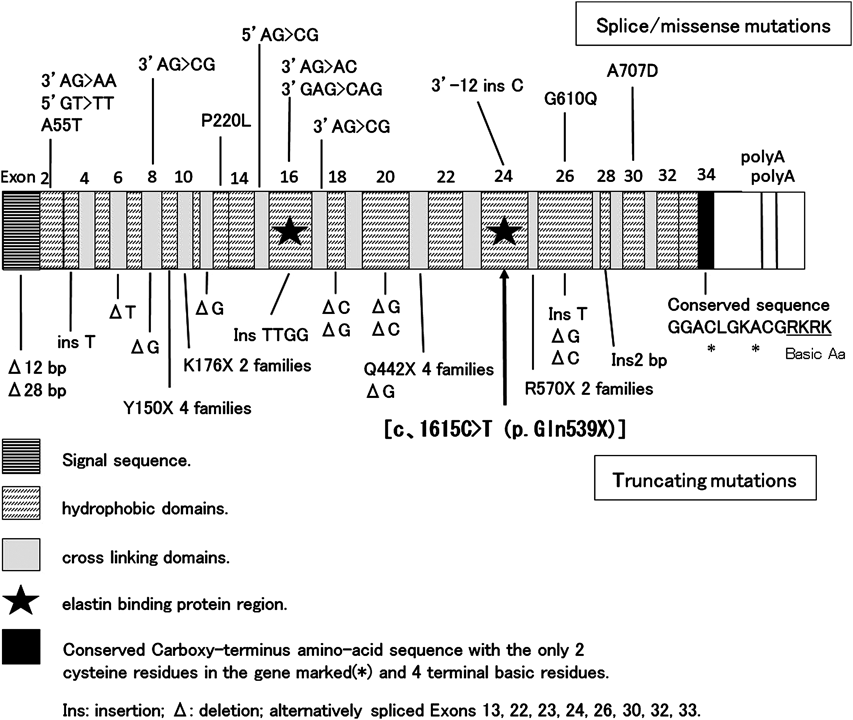
As SVAS and PPS were the characteristic cardiovascular lesions of WBS, we tried to detect a deletion at chromosome 7q11.23 by FISH (Elastin probe from Vysis), but no deletion was detected. So, we obtained informed consent from her parents and analyzed ELN which was responsible for cardiovascular lesions of WBS. We got approval for gene analysis from Toyama University Ethics Committee. We amplified the Exon coding region and the contiguous region including the introns for ELN by using the primer which was obtained from her peripheral blood lymphocyte genome DNA (we are ready to disclose the primer information when you need). We purified the amprified products by QIAquick polymerase chain reaction (PCR) purification method (QIAGEN, Hilden, Germany) and analyzed mutations by direct-sequencing using ABI 3130xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA). As a result, we identified the nonsense mutation in heterozygote of ELN [c.1615C>T (p.Gln539X)] (Fig. 3). Using direct-sequencing, we also analyzed PTPN11, RAF1, SOS1, Shoc2, PKA and PKC, which were responsible for Noonan syndrome having PS with high probability, but no mutations were detected. We have not analyzed the mutations of her parents and brothers yet.
WBS is a contiguous gene syndrome according to a partial deletion at chromosome 7q11.23. A cardiovascular lesions of WBS were SVAS, PPS, aortic hypoplasia, coarctation of aorta and mitral valve prolapse.2) SVAS and PPS are especially characteristic for WBS. As sporadic SVAS is rare, estimated to be 5–7: 100000, so we need to take WBS into account in case of diagnosing SVAS.3) ELN is identified as a responsible gene for cardiovascular lesions of WBS.
ELN contribute to expression of the elastin. The elastin is a main component of elastic fiber and is widely distributed in artery, especially in a root of aorta, skin and lungs. It is advocated that decreasing of the elastin lead to increasing of the collagen in tunica media of artery and vessel wall becomes thicken and harden. In ELN gene knockout mice, it was reported that a homozygous mouse died in a few days according to aortic obstruction with hypertrophy of smooth muscle and a heterozygous mouse had a half expression of an elastin protein and messenger RNA.4) A point mutation of ELN can occur cardiovascular lesions.5) Frangiskakis and Tassabehji report the plural familial SVAS families with ELN mutation, so it is considered that ELN is also responsible gene for cardiovascular lesions of familial SVAS.6, 7) Cardiovascular lesions can occur not only for a great mutation, including nonsense mutation and frameshift mutation, but also for a missense mutation, and the expression pattern is variable.5, 7–9)
We present the case with a nonsense mutation in heterozygote of ELN [c.1615C>T (p.Gln539X)], that has never been reported before. Among ELN mutations, many cases with missense mutation have relatively mild cardiovascular lesions and many cases with nonsense mutation or frameshift mutation have severe cardiovascular lesions. In addition, since Exon 16 and Exon 24 contain elastin binding protein region and severe cardiovascular lesions (SVAS, PPS, aortic hypoplasia) are observed in cases with Exon 16 mutation (frame shift), it is suggested that a deletion at Exon 16 or Exon 24 tends to cause more severe cardiovascular lesions. In this case, in addition to SVAS, PPS and supravalvular pulmonary stenosis, bilateral renal artery stenosis was also observed, and renal artery stenosis is rare in the past reports. A nonsense-mediated RNA decay is also considered for nonsense mutation, but in this case, because of nonsense mutation of Exon 24 which has an important role among ELN genes, it is considered that serious quantitative or qualitative change of elastin were caused and cardiovascular lesions characteristic of WBS and other widespread cardiovascular lesions developed by this ELN mutation.9)
In this case, the limitations is as follows; the follow-up period is short in one year, so it is insufficient to examine other lesions except for cardiovascular lesions and development. Second, it is not directly proven that ELN mutation is causing functional abnormality of elastin.
We presented the case having characteristics cardiovascular lesions of WBS with a nonsense mutation in ELN that has never been reported before. In cases with characteristic cardiovascular lesions of WBS, we need to consider the case having only ELN mutation with FISH negative and understand for pathological condition of cardiovascular lesion and extracardial lesion.
Conflicts of Interest
The authors declare no conflict of interest.
Authorship Contribution
S.T. designed and wrote the paper. S.T., H.N., K.I., S.O., K.H. and Y.A. practiced medicine. S.H. transferred the patient to our hospital and provided the information at birth. T.N. performed gene analysis. F.I. was responsible for the paper and gave a critical review on intellectual content.
Originally published in Journal of Japanese Pediatric Society, Vol. 121 (2017) No. 7, pp. 1179–1183
引用文献References
1) Strømme P, Bjømstad PG, Ramstad K: Prevalence estimation of Williams syndrome. J Child Neurol 2002; 17: 269–271
2) Wessel A, Pankau R, Kececioglu D, et al: Three decades of follow-up of aortic and pulmonary vascular lesions in Williams Beuren syndrome. Am J Med Genet 1994; 52: 297–301
3) Matsumoto K, Kawai H: Left ventricular outflow tract anomaly and aortic abnormality. J Clin Echocardiograph 2012; 13: 236–241 (in Japanese)
4) Li DY, Faury G, Taylor DG, et al: Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest 1998; 102: 1783–1787
5) Delio M, Pope K, Wang T, et al: Spectrum of elastin sequence variants and cardiovascular phenotypes in 49 patients with Williams–Beuren syndrome. Am J Med Genet 2013; 161A: 527–533
6) Frangiskakis JM, Ewart AK, Morris CA, et al: LIM-kinase 1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell 1996; 86: 59–69
7) Tassabehji M, Metcalfe K, Donnai D, et al: Elastin: Genomic structure and point mutation in patients with supravascular aortic stenosis. Hum Mol Genet 1997; 6: 1029–1036
8) Li DY, Toland AE, Boak BB, et al: Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Hum Mol Genet 1997; 6: 1021–1028
9) Metcalfe K, Rucka AK, Smoot L, et al: Elastin: Mutational spectrum in supravascular aortic stenosis. Eur J Hum Genet 2000; 8: 955–963