Primary cardiac tumors are rare in pediatric population with extremely low prevalence. The majority of such tumors are benign, while approximately 10% are malignant. Rhabdomyoma is the most common cardiac tumor in children seen in more than 60% of all primary cardiac tumors.1–3) Cardiac rhabdomyomas tend to regress over time, and spontaneous remission is frequently the case in children. This histological type of the cardiac tumor is commonly associated with tuberous sclerosis complex (TSC).4) Recently, mammalian target of rapamycin (mTOR) inhibitors have been reported for treating symptomatic cardiac rhabdomyoma in infants. This review provides a brief overview of this entity of disease summarizing information available, in addition to current data regarding mTOR inhibitors in the treatment of pediatric cardiac rhabdomyoma with TSC; the agent may be used in the clinical practice.
The physician should assess a cardiac tumor based on its size, shape, site of attachment, and the area of extension. Echocardiography is an imaging modality commonly used for non-invasive screening and evaluation of a cardiac mass in children, although echocardiography cannot provide the histopathological information. Cardiac magnetic resonance imaging (MRI) can provide further assessment of the tumor, including anatomical information, especially useful for the decision making process towards surgical resection. While cardiac MRI is a preferred modality, it may have potential risks associated with sedation or general anesthesia during examination. Computed tomography (CT) can also play an important role in evaluation for a cardiac mass, providing anatomical information such as calcification, and comprehensive assessment of metastatic disease. Because the majority of primary cardiac tumors in children are benign, CT has special reservations about the potential for increased radiation exposure.
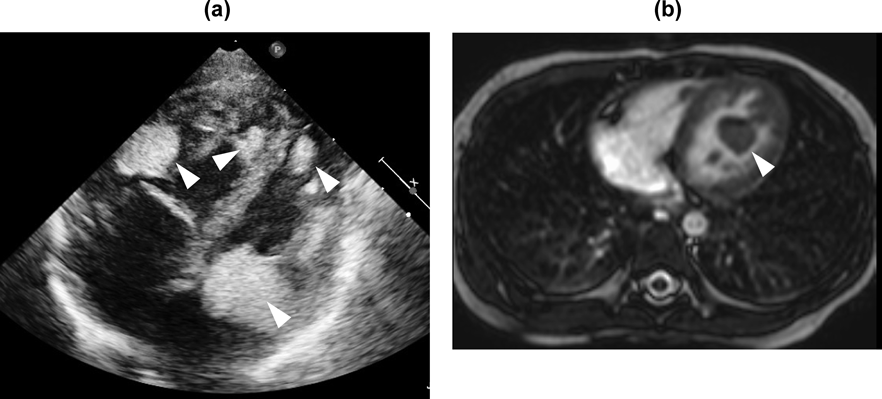
Table 1 shows the imaging characteristics of primary cardiac tumors in children. Cardiac rhabdomyomas are commonly multiple (in more than 60% of patients) and multiple rhabdomyomas are highly associated with TSC.5) Cardiac rhabdomyomas are located on either of the ventricles, especially on the left, with or without outflow obstruction. Echocardiography can detect rhabdomyomas as hyperechoic solid masses (Fig. 1a). CT provides anatomical information such as the myocardium contiguous with the tumors, or the nature of the tissues of the tumors themselves. This modality is helpful, as mentioned above, for comprehensive assessment of metastatic disease.6) Cardiac MRI shows isointense to myocardium on T1-weighted images and hyperintense on T2-weighted images (Fig. 1b).7) With contrast-enhanced imaging, cardiac rhabdomyomas appear as areas weakly enhanced. CT and cardiac MRI are non-invasive diagnostic tools. The imaging features from these modalities, although non-specific, are often used to characterize the mass further, eventually facilitating diagnoses and guiding treatments.6, 7) Histological assessment remains as a gold standard for confirmation of a pathological diagnosis.
Table 1 Echocardiographic and cardiac MRI features of cardiac tumors in children| Tumor | Location | Echocardiography | Cardiac MRI |
|---|
| Rhabdomyoma | Ventricle (LV>RV)
multiple>solitary | contiguous with myocardium
well-circumscribed hyperechoic mass | isointense to myocardium (T1)
hyperintense to myocardium (T2) |
| Fibroma | Ventricle (LV free wall)
solitary>multiple | heterogenous mass
calcifications | non-contractile mass
delayed enhancement |
| Teratoma | intrapericardial
solitary>multiple | cystic and solid mass
pericardial effusion | hypointense |
| Myxoma | atrium (LA>RA)
attachment to interatrial wall
solitary>multiple | mobile and heterogeneous mass
irregular borders | hyperintense to myocardium (T2)
heterogeneous enhancement |
| LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. |
Most children with cardiac rhabdomyoma are usually asymptomatic; some patients have heart failure or arrhythmias.1–3) Presentation of such clinical symptoms depends on the size and location of the tumor. After birth, clinical symptoms are related to obstruction across the inflow or outflow tracts of the ventricles. Outflow tract obstruction is a potential risk of cardiac dysfunction due to impairment of coronary perfusion, lethal ventricular arrhythmia, and sudden cardiac death.8–10) A huge cardiac tumor can cause left atrioventricular valve stenosis and its hemodynamics resembles to hypoplastic left heart syndrome during the neonatal period. Cardiac rhabdomyoma may be noted coincidentally with cardiac arrhythmia. The incidence of arrhythmias had been reported to be 16 to 47% of cases11–13) in children. Wolff-Parkinson-White syndrome in children with cardiac rhabdomyoma were ten times more frequent than those in the general population (1.5% vs 0.15%).14) This suggests that tumor cells may create continuity through the atrioventricular junction. As the cardiac tumor regresses, this arrhythmia results in spontaneous resolution. The majority of patients having cardiac rhabdomyoma and Wolff-Parkinson-White syndrome were asymptomatic. Among all arrhythmias, ventricular tachycardia is commonest (6%) in children with cardiac rhabdomyoma.15) Ventricular arrhythmia is a potential risk of sudden cardiac death, but arrhythmic death is rare in this setting.
Approximately 80% of cardiac rhabdomyomas spontaneously regress and morbidity and mortality by the lesion remain low.16, 17) Because prognosis of cardiac rhabdomyoma in children is commonly favorable, active treatments for the tumor masses should be performed only when symptomatic and life-threatening. The symptoms and the prognosis depend on the size and location of the tumor, thus these features should be monitored on echocardiography during follow-up. Less commonly, cardiac rhabdomyomas may grow or newly appear around 10–15 years old. The natural history of cardiac rhabdomyoma in adolescents is possibly different from those in younger children. After spontaneous regression of a tumor, cardiac rhabdomyoma may regrow in puberty, especially in young girls.18) Rhabdomyoma can develop under the endocardium or the epicardium. A tumor in the sub-epicardial layer can stretch the coronary artery when greater than 2 cm in diameter, even without hemodynamic compromise.19)
Cardiac Rhabdomyomas Associated with TSC
Cardiac rhabdomyomas are found in approximately 50% of patients with TSC; in turn, more than 60% of cardiac rhabdomyomas are associated with TSC. TSC is inherited in an autosomal dominant trait and have mutations in either of the tumor-suppressor genes TSC1 (9q34) or TSC2 (16p14.3). The TSC protein complex inhibits mechanistic target of rapamycin complex 1, which controls cell growth, proliferation, autophagy, and protein and lipid synthesis. Mutations in the TSC1 and TSC2 result in hyperactivation of the mTOR signaling pathway, and subsequently resulting in the development of tumor-like lesions called hamartomas.5) Cardiac rhabdomyomas were more frequent in patients with mutation of TSC2 than those of TSC1 mutation.3) In addition, the clinical features of TSC include malformations of subependymal giant cell astrocytomas (brain tumor), renal angiomyolipomas, facial angiofibromas, and pulmonary lymphangioleiomyomatosis. The prognosis is highly variable and depends on the severity of symptoms in TSC patients. Life-threatening conditions related to brain tumors, renal or pulmonary lesions, and, in general, cardiac rhabdomyomas, do not affect prognosis in children with TSC. Cardiac rhabdomyomas are usually larger in patients with TSC than in those without.3) Multiple tumors are common association with TSC, causing outflow tract obstruction more frequently. Therefore, genetic testing may be considered after birth when tumors are multiple.
Although genetic testing is not required in every patient with TSC, the test is useful to confirm or rule out the circumstance in patients with cardiac tumor in whom TSC is suspected but diagnostic criteria do not entirely meet it. Genetic counseling can help the family to make informed decisions about genetic testing, especially when the child is already diagnosed with TSC and the parents wish to have more children in the future because the condition has 95% penetrance.3, 4) The mutations are spontaneous in 60% of patients while the remaining are familial. TSC1 gene variants are more common in familial cases of TSC, while TSC2 gene variants occur more in sporadic cases. Patients with TSC2 gene variants have severer forms of TSC than those in TSC1 gene variants.3, 4) After birth, the diagnosis of cardiac tumors can be established in symptomatic patients. Non-invasive test such as echocardiography is a useful diagnostic tool for assessment of cardiac tumors. Diagnosis of TSC is based on updated international criteria. Still, early diagnosis of TSC is a challenge.20) A definite diagnosis of TSC is made when a patient has either 2 major features such as hypomelanotic macules and cardiac rhabdomyoma, or 1 major with 2 minor features such as confetti skin lesions. In general, the first sign may be seizures or white patches on the skin. When TSC is suspected, it may be possible to diagnose the condition with a careful examination of clinical manifestations, even in early infancy.
Fetal cardiac rhabdomyoma can be detected on 20 weeks of gestation and some cases are diagnosed at the end of the third trimester. Many tumors of this type grow until 32 weeks of gestational age, and eventually get smaller during the first year of life.21) This suggested that hormonal stimulation in utero were associated with tumor growth.22) Cardiac rhabdomyoma may present during the prenatal period as fetal arrhythmia, heart failure, or hydrops. Prognosis of fetal hydrops is poor with 4 to 6% risk of stillbirth.23, 24) A previous study demonstrated factors leading to difference in clinical outcome between those with cardiac rhabdomyoma prenatally diagnosed and postnatally diagnosed.24) Twenty fetuses were diagnosed at 28 weeks of gestational age. Out of these twenty, one died in utero, and 18 were delivered at term. Although none had prenatal hemodynamic complications, 7 (37%) required medical treatments or surgical intervention after birth. On the other hand, 26 patients were postnatally diagnosed with cardiac rhabdomyoma. The incidences of cardiac symptoms and TSC were not different between the groups of patients having prenatal and postnatal diagnosis. This study suggested that cardiac rhabdomyoma are typically well tolerated with a low risk of fetal death, and that natural prognosis of rhabdomyoma prenatally detected is mostly favorable. In some cases, however, cardiac tumor may develop during the fetal period and after birth, resulting in reduced cardiac output due to intracardiac obstruction.21, 23, 24) Thus, the physician should monitor the space-occupying tumor in fetuses and children.
1. Surgical Resection
Since cardiac rhabdomyomas have a potential of spontaneous regression, follow-up without surgical procedures is sensible in children. Indication for surgery is only against severe obstruction across the ventricular outflow/inflow or lethal arrhythmias. A previous study from the Hospital for Sick Children in Toronto documented outcomes of primary cardiac tumors located at the right and left ventricular outflow tracts.10) Of 130 children in their series, 36 (28%) had a cardiac tumor at either of the outflow tracts, with rhabdomyoma being seen in 26 (72%). Fourteen out of 36 children had a peak pressure gradient >20 mmHg across the outflow tract. Twelve out of 36 (33%) underwent surgical repair. These patients tended to be sicker clinically than those in whom clinical course was conservatively observed. One male patient died at 1 year old of acute obstruction across the left ventricular outflow tract by a rhabdomyoma. This study suggested that surgical resection of tumors should be considered for symptomatic patients with significant hemodynamic impediments.
A multi-center study from European countries reported clinical outcomes in 89 children undergoing surgery for cardiac tumors (a median age 4 months old, with a range from 1 day to 18 years), including 32 rhabdomyoma (36%).25) Complete resection was achieved in 62 (70%) patients, partial resection in 21 (24%), and cardiac transplant in 4 (4.5%). There were no differences in survival and adverse events after complete and partial resection in benign tumors. This retrospective registry revealed surgery for primary cardiac tumors had a favorable outcome. According to no recurrence after partial resection, surgery is one of effective strategies for treating rhabdomyomas. Other retrospective studies regarding surgical resection of cardiac tumors in children demonstrated low mortality rate, especially for cardiac rhabdomyoma.26–28) Based on these previous data, resection of tumors in the pediatric population is associated with improvement of symptoms and good outcomes. This is particularly true in patients with severe obstruction presenting hemodynamic compromise and refractory arrhythmias in whom surgical intervention is pertinently indicated.
Surgical resection of a large cardiac rhabdomyoma, however, is somewhat concerning, associated with lethal complications such as arrhythmia and sudden death. Moreover, a surgical technique through the aortic valve to approach the tumor has been used for cardiac rhabdomyoma causing left ventricular outflow obstruction.26) The retrograde approach is still challenging because the intracardiac maneuver through the aortic valve is limited by the size of the aortic annulus. The tumor should be removed in several pieces to avoid damage to the aortic valve. Aortic valve insufficiency should not be induced during the procedure, and that is why left ventricular outflow obstruction remains as a surgical challenge.29)
2. Inhibitors of mTOR
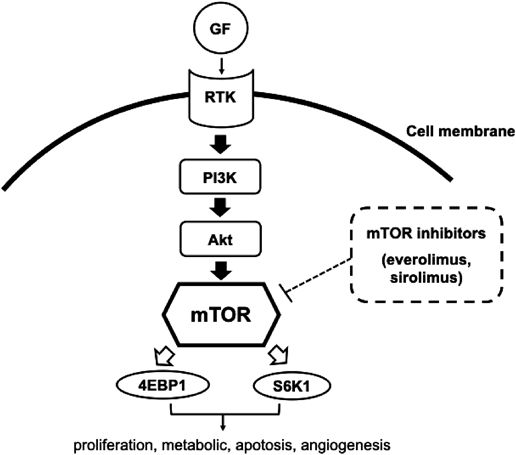
Inhibitors of mTOR, such as Everolimus or Sirolimus, are molecularly targeted drugs. mTOR is a serine/threonine kinase which controls cell growth, proliferation, metabolism, apoptosis, and angiogenesis (Fig. 2). Children with TSC1 or TSC2 mutations have hyperactivation of the mTOR pathway, leading to abnormal cell proliferation including hamartoma. Only Everolimus is currently approved, in the United States and Japan, for treatment of subependymal giant cell astrocytomas and renal angiomyolipomas. mTOR inhibitors have been investigated for treating pediatric patients with TSC, and are also potential therapeutic agents for cardiac rhabdomyoma in TSC.30–35) A systematic review demonstrated that use of mTOR inhibitors (Everolimus and Sirolimus) in children with TSC and cardiac rhabdomyomas was effective and safe.36) In total, 41 patients were included (from 0 to 18 years old). Most patients had mTOR inhibitors administrated during the neonatal period (71%) or infancy (19.5%). Duration of the treatment was reportedly from 28 to 390 days (a median 70 days). The initial dose for Everolimus was 4.5 mg/m2/week or 0.05 to 1 mg/day, and that for Sirolimus 1 mg/m2/day or 0.25 to 0.5 mg/day, although overall treatment doses were various in these studies. Serum levels of Everolimus and Sirolimus were targeted around 5–15 ng/mL. According to the report, as a whole, the size of cardiac rhabdomyoma was reduced in 95%, and clinical condition improved in 91%. The 34 adverse events were noted in 33 patients, their severity being mild in the majority. Table 2 showed the characteristics of children treated with Everolimus and Sirolimus from published studies. These are non-randomized data, and lack of targeted doses and serum levels is obvious. Because of the preliminary nature, use of mTOR inhibitors should not be widely justified for treatment of cardiac rhabdomyoma. In addition, safety profile is still a major concern especially in premature neonate, because long-term adverse effects remain unknown. Therefore, this treatment protocol should be applied in selected patients. Currently, a placebo-controlled, double-blind phase II trial (ORACLE; Everolimus for cardiac rhabdomyomas in tuberous sclerosis) is conducted. This study has been started as the first randomized clinical trial assessing the efficacy of Everolimus as a specific therapy for symptomatic cardiac rhabdomyoma in children with TSC.37)
Table 2 Summary of treatments with mTOR inhibitors| mTOR inhibitor | Number of patients | Age | Initial dose | Serum levels | Efficacy* | Reference number |
|---|
| Everolimus | 4 | 2–20 days | 0.1 mg/day | 5–15 ng/mL | Yes | 32 |
| 1 | neonate | 0.1 mg/day | 8 ng/mL | Yes | 33 |
| 1 | 36 days | 0.5 mg/day | not available | Yes | 34 |
| Sirolimus | 1 | 3 days | 0.25 mg/day | 69.7 ng/mL | Yes | 35 |
| 1 | 18 days | 0.25 mg/day | 42.1 ng/mL | Yes | 36 |
| 1 | 10 days | 0.5 mg/day | 26 ng/mL | Yes | 37 |
| * Efficacy: size of tumor reduced or no clinical deterioration. |
Clinical prognosis with cardiac rhabdomyoma is generally good owing to the spontaneous reduction of its size. Surgical resection of tumors should be considered to relieve mechanical obstruction. Efficacy and safety of mTOR inhibitors in children have been reported in case studies. A randomized, placebo-controlled study with TSC related rhabdomyoma is currently conducted. Nonetheless, many cardiac rhabdomyomas may regress during conservative follow-up. Thus, it is difficult to evaluate an obvious role of mTOR inhibitors in pediatric rhabdomyoma. For the time being, observational studies are informative for assessing the benefit and the risk of treatment strategies in children. In the future, a national registry for cardiac rhabdomyoma in children should be organized at Japanese institutions.
Financial Support
This research has not received any specific grant from any funding agency, commercial, or not-for-profit sectors.
Conflicts of Interest
None.
Author Contributions
All authors have contributed to the drafting of this manuscript and have approved the submitted version.
引用文献References
1) Simcha A, Wells BG, Tynan MJ, et al: Primary cardiac tumors in childhood. Arch Dis Child 1971; 46: 508–514
2) Becker AE: Primary heart tumors in the pediatric age group: A review of salient pathologic features relevant for clinicians. Pediatr Cardiol 2000; 21: 317–323
3) Madueme P, Hinton R: Tuberous sclerosis and cardiac rhabdomyomas: A case report and review of the literature. Congenit Heart Dis 2011; 6: 183–187
4) Harding CO, Pagon RA: Incidence of tuberous sclerosis in patients with cardiac rhabdomyoma. Am J Med Genet 1990; 37: 443–446
5) Tworetzky W, McElhinney DB, Margossian R, et al: Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol 2003; 92: 487–489
6) Tao TY, Yahyavi-Firouz-Abadi N, Singh GK, et al: Pediatric cardiac tumors: Clinical and imaging features. Radiographics 2014; 34: 1031–1046
7) Sparrow PJ, Kurian JB, Jones TR, et al: MR imaging of cardiac tumors. Radiographics 2005; 25: 1255–1276
8) Behram M, Oğlak SC, Acar Z, et al: Fetal cardiac tumors: Prenatal diagnosis, management, and prognosis in 18 cases. J Turk Ger Gynecol Assoc 2020; 21: 255–259
9) Gupta A, Narula N, Mahajan R, et al: Sudden death of a young child due to cardiac rhabdomyoma. Pediatr Cardiol 2010; 31: 894–896
10) Nield LE, Mendelson M, Ahmad N, et al: Clinical review of obstructive primary cardiac tumors in childhood. Congenit Heart Dis 2014; 9: 244–251
11) Mas C, Penny DJ, Menahem S: Pre-excitation syndrome secondary to cardiac rhabdomyomas in tuberous sclerosis. J Paediatr Child Health 2000; 36: 84–86
12) Kusano KF, Ohe T: Cardiac tumors that cause arrhythmias. Card Electrophysiol Rev 2002; 6: 174–177
13) Beghetti M, Gow RM, Haney I, et al: Pediatric primary benign cardiac tumors: A 15-year review. Am Heart J 1997; 134: 1107–1114
14) O’Callaghan FJ, Clarke AC, Joffe H, et al: Tuberous sclerosis complex and Wolff-Parkinson-White syndrome. Arch Dis Child 1998; 78: 159–162
15) Miyake CY, Del Nido PJ, Alexander ME, et al: Cardiac tumors and associated arrhythmias in pediatric patients, with observations on surgical therapy for ventricular tachycardia. J Am Coll Cardiol 2011; 58: 1903–1909
16) Bosi G, Linthermans JP, Pellegrino PA, et al: The natural history of cardiac rhabdomyoma with and without tuberous sclerosis. Acta Paediatr 1996; 85: 928–931
17) Smythe JF, Dyck JD, Smallhorn JF, et al: Natural history of cardiac rhabdomyoma in infancy and childhood. Am J Cardiol 1990; 66: 1247–1249
18) Jóźwiak S, Kotulska K, Kasprzyk-Obara J, et al: Clinical and genotype studies of cardiac tumors in 154 patients with tuberous sclerosis complex. Pediatrics 2006; 118: e1146–e1151
19) Chao AS, Chao A, Wnag TH, et al: Outcome of antenatally diagnosed cardiac rhabdomyoma: Case series and a meta-analysis. Ultrasound Obstet Gynecol 2008; 31: 289–295
20) Northrup H, Aronow ME, Bebin EM, et al: Behalf of the International Tuberous Sclerosis Complex Consensus Group: Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatr Neurol 2021; 123: 50–66
21) Fesslova V, Villa L, Rizzuti T, et al: Natural history and long-term outcome of cardiac rhabdomyomas detected prenatally. Prenat Diagn 2004; 24: 241e8
22) Nir A, Ekstein S, Nadjari M, et al: Rhabdomyoma in the fetus: Illustration of tumor growth during the second half of gestation. Pediatr Cardiol 2001; 22: 515–518
23) Holley DG, Martin GR, Brenner JI, et al: Diagnosis and management of fetal cardiac tumors: A multicenter experience and review of published reports. J Am Coll Cardiol 1995; 26: 516e20
24) Bader RS, Chitayat D, Kelly E, et al: Fetal rhabdomyoma: prenatal diagnosis, clinical outcome, and incidence of associated tuberous sclerosis complex. J Pediatr 2003; 143: 620–624
25) Padalino MA, Vida VL, Boccuzzo G, et al: Surgery for primary cardiac tumors in children: Early and late results in a multicenter European Congenital Heart Surgeons Association study. Circulation 2012; 126: 22–30
26) Pipitone S, Mongiovì M, Grillo R, et al: Cardiac rhabdomyoma in intrauterine life: Clinical features and natural history. A case series and review of published reports. Ital Heart J 2002; 3: 48–52
27) Centofanti P, Di Rosa E, Deorsola L, et al: Primary cardiac neoplasms: Early and late results of surgical treatment in 91 patients. Ann Thorac Surg 1999; 68: 1236–1241
28) Bielefeld KJ, Moller JH: Cardiac tumors in infants and children: Study of 120 operated patients. Pediatr Cardiol 2013; 34: 125–128
29) Black MD, Kadletz M, Smallhorn JF, et al: Cardiac rhabdomyomas and obstructive left heart disease: Histologically but not functionally benign. Ann Thorac Surg 1998; 65: 1388–1390
30) Demir HA, Ekici F, Erdem AY, et al: A challenging drug in the treatment of multifocal inoperable cardiac rhabdomyoma. Pediatrics 2012; 130: e243–e247
31) Fatou AW, Goyer I, Raboisson MJ, et al: Accelerated cardiac rhabdomyoma regression with everolimus in infants with tuberous sclerosis complex. Pediatr Cardiol 2017; 38: 394–400
32) Martínez-García A, Michel-Macías C, Cordero-González G, et al: Giant left ventricular rhabdomyoma treated successfully with everolimus: Case report and review of literature. Cardiol Young 2018; 28: 903–909
33) Lawley C, Popat H, Wong M, et al: Dramatic response to sirolimus therapy in a premature infant with massive cardiac rhabdomyoma. JACC Case Rep 2019; 1: 327–331
34) Lee SJ, Song ES, Cho HJ, et al: Rapid regression of obstructive cardiac rhabdomyoma in a preterm neonate after sirolimus therapy. Biomed Hub 2017; 2: 1–6
35) Breathnach C, Pears J, Franklin O, et al: Rapid regression of left ventricular outflow tract rhabdomyoma after sirolimus therapy. Pediatrics 2014; 134: e1199–e1202
36) Sugalska M, Tomik A, Jóźwiak S, et al: Treatment of cardiac rhabdomyomas with mTOR inhibitors in children with tuberous sclerosis complex-a systematic review. J Environ Res Public Health 2021; 18: 4907
37) Stelmaszewski EV, Parente DB, Farina A, et al: Everolimus for cardiac rhabdomyomas in children with tuberous sclerosis. The ORACLE study protocol (everOlimus for caRdiac rhAbdomyomas in tuberous sCLErosis): A randomised, multicentre, placebo-controlled, double-blind phase II trial. Cardiol Young 2020; 30: 337–345
 1,Ryo Inuzuka2,Tohru Kobayashi3,Tomomi Ueda4,Jun Maeda5,Yoshiyuki Furutani6,Kei Inai6,Mitsuhiro Kamisago7,Hiroyuki Yamagishi8Shinichi Takatsuki
1,Ryo Inuzuka2,Tohru Kobayashi3,Tomomi Ueda4,Jun Maeda5,Yoshiyuki Furutani6,Kei Inai6,Mitsuhiro Kamisago7,Hiroyuki Yamagishi8Shinichi Takatsuki