Anomalous origin of a unilateral pulmonary artery from the ascending aorta (AAo) is a rare congenital malformation, which is characterized by the right or the left pulmonary artery originating from the AAo with separate semilunar valves. The contralateral pulmonary artery is normally connected to the pulmonary trunk (PT) arising from the right ventricle. It accounts for 0.1% of all congenital heart diseases.1) The anomalous pulmonary artery is more frequently on the right than left.1, 2) The anomalous pulmonary artery does not usually have a direct connection to the PT. Only three cases have been reported with continuity between the anomalous right pulmonary artery (RPA) and the PT.3–5) Their morphological diagnosis is still controversial, whether anomalous origin of the RPA from the AAo (AORPA) or aorto-pulmonary window (APW).6) We herein report a premature baby with AORPA or APW whose RPA communicating with the PT via a small lumen.
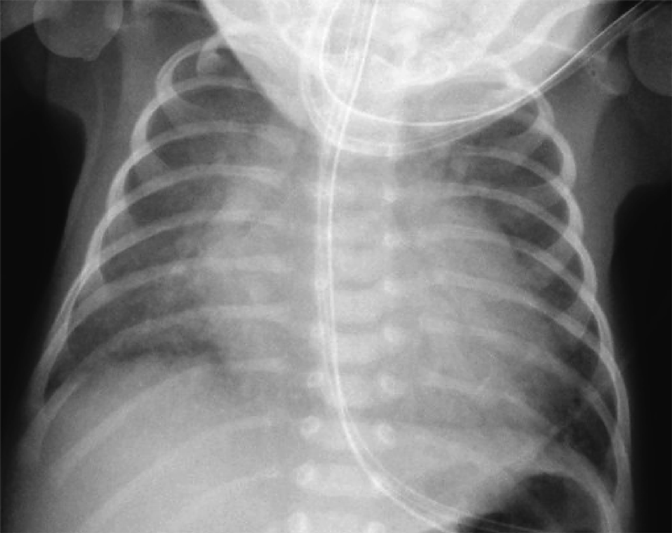
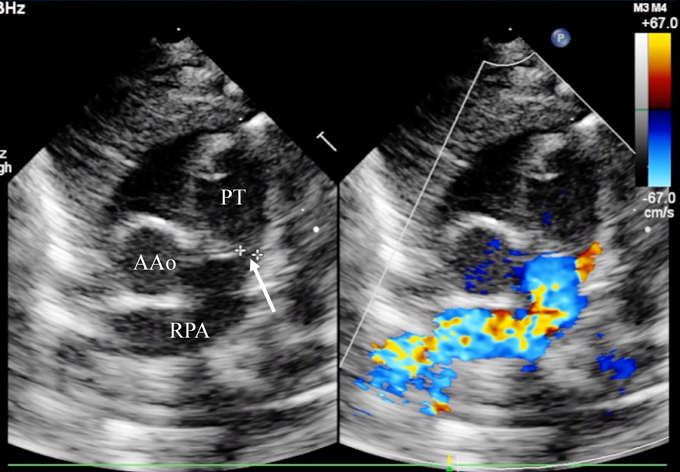
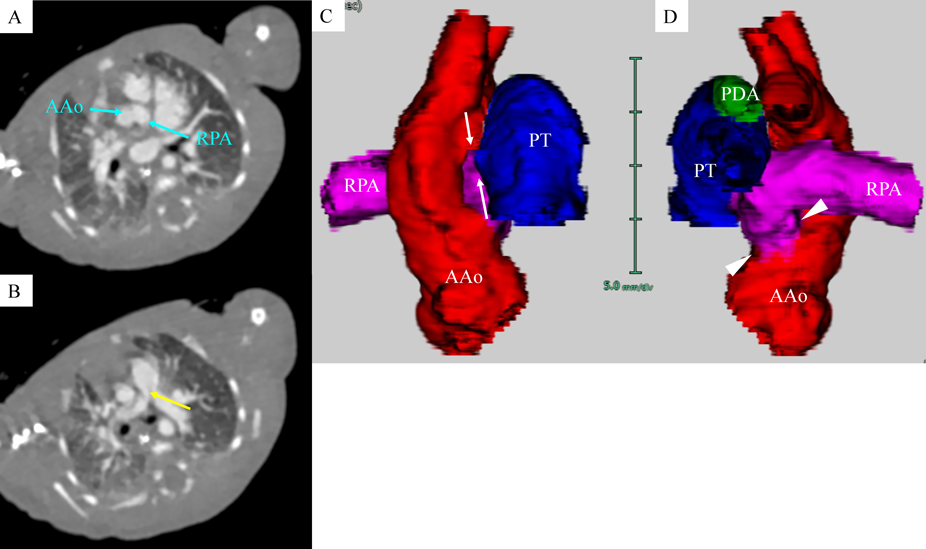
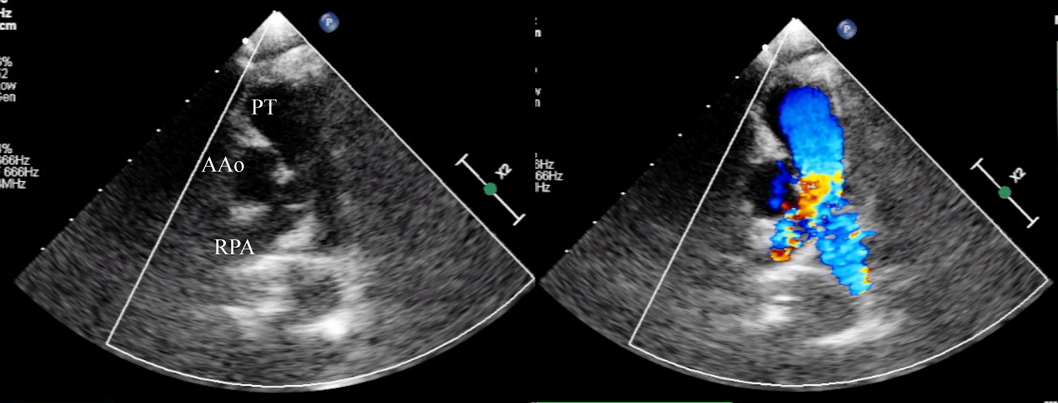
A female baby was delivered by an emergent cesarean section secondary to fetal bradycardia at a gestation of 33 weeks and 4 days. She weighed 1,700 g, and was eventually referred to our hospital at 12 days old. During pregnancy, her mother was doing well without detection of any congenital anomaly including heart disease on routine fetal ultrasonograms. She had no family history regarding congenital heart disease. She was suffering from progressive respiratory distress and congestive heart failure. Levine 2/6 continuous murmur was heard at the left sternal border of the second intercostal space. Her heart rate, respiratory rate and blood pressure were 164 beats/min, 70 breaths/min and 60/30 mmHg, respectively. Oxygen saturation was 99% on room air. Chest X-ray showed cardiomegaly with right pulmonary plethora (Fig. 1). An electrocardiogram showed biatrial overload. Her serum B-type natriuretic peptide level was 3,383 pg/mL. The RPA originated from the left posterior aspect of the proximal AAo. The orifice of the RPA at the proximal AAo was 5.0 mm. Moreover, the RPA was connected to the PT via a 2.0 mm-diameter lumen at the intrapericardial portion adjacent to its aortic origin. She was diagnosed as having AORPA, left aortic arch, patent ductus arteriosus bridging normally between the aortic arch and the PT, and patent fossa ovale by transthoracic echocardiography and computed tomography (Figs. 2, 3). Nitrogen inhalation therapy was started to treat high pulmonary flow.
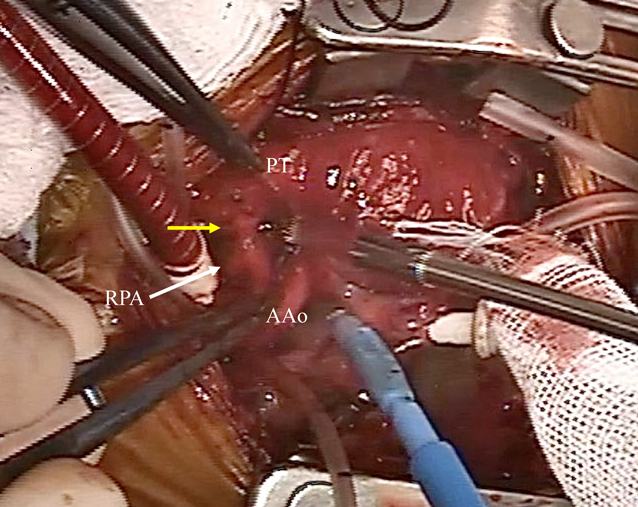
Surgical treatment was performed at 13 days of age. The heart was exposed through a median full sternotomy. The RPA originated from the left posterior aspect of the AAo just above the sinus of Valsalva, and was connected to the PT (Fig. 4) as shown preoperatively. The RPA was dissected and snared between the superior vena cava and the AAo. Cardiopulmonary bypass was established via cannulations into the distal AAo and the right atrium. The patent ductus arteriosus was ligated and divided. The connection between the RPA and the PT was 3 mm in external diameter. After cardioplegic arrest, the RPA was detached from the AAo and the PT. The incised wall of the AAo was directly oversewn. The RPA was reimplanted to the PT, also directly, using 7-0 Prolene® (Johnson & Johnson Services, Inc., New Brunswick, New Jersey, USA) running suture. Weaning from cardiopulmonary bypass was smooth. Cardiopulmonary bypass time and aortic cross-clamp time were 89 and 35 minutes, respectively. Pulmonary arterial and aortic pressure was 26/18 (22) and 85/49 (63) mm Hg, respectively. Primary sternal closure was achieved. The postoperative course was uneventful. She was discharged on the postoperative day 39 (body weight of 2,146 g). She is asymptomatic with normal growth. The RPA showed no stenosis or deformity at five years postoperatively (Fig. 5).
AORPA has two patterns morphologically: the proximal and the distal types.1, 2) The former is characterized by an anomalous RPA that usually arises from the posterior or right lateral aspect of the AAo close to the aortic valve. Its orifice is widely open in the majority. As for the distal type, the morphological feature is the anomalous RPA usually originating from the AAo close to the brachiocephalic artery, or much less frequently from the brachiocephalic artery. Its orifice is likely surrounded by a ductal tissue, and becomes stenotic in many instances.1) AORPA is often associated with patent ductus arteriosus, bridging between the aortic arch and the PT as usual. Occasionally, an aortopulmonary septal defect or interrupted aortic arch coexists.
In embryogenesis, the AAo and the PT separate from the initial aortic sac. Also, the right sixth pharyngeal arch migrates leftward and fuses to the left sixth pharyngeal arch, forming the distal part of the PT. It is hypothesized that the proximal type of AORPA is a result of insufficient leftward migration of the right sixth arch and imbalanced septation of the aortic sac.7) The RPA is rarely in continuity with the PT. To the best of our knowledge, only three reports have described continuity between the RPA and the PT.3–5) These cases did not have luminal continuity. Our case had communication through a small lumen between the RPA and the PT, which has never been reported. The proximal RPA was clearly identified as seen in the previous cases.3–5) The orifice of the RPA at the proximal AAo was much greater than the additional channel between the RPA and the PT. We diagnosed her malformation, therefore, as unusual form of AORPA. An alternative diagnosis may be AORPA together with APW. Ho et al. reported that these abnormalities can coexist.6) The present case can sit within this spectrum of morphological variations. Somebody would argue that the diagnostic entity remains controversial. We contemplate that imbalanced and incomplete septation of the aortic sac could have resulted in the unusual pathway between the RPA and the PT.5)
Congestive heart failure with high pulmonary flow usually progresses rapidly in patients with the proximal type of AORPA. Severe pulmonary hypertension and pulmonary vascular obstructive disease also develop early in life.8) In this respect, early surgical repair is pertinent, even during the neonatal period.2) The result of palliative surgery was disappointing.2) Direct anastomosis of the RPA to the PT is recommended as far as feasible.2, 9, 10) Nonetheless, the distance between the mobilized RPA and the PT varied. Stretching and/or kinking of the reconstructed RPA should be avoided in terms of future stenosis. When direct reimplantation was unapplicable, some surgical techniques have been described, such as use of a flap of the aortic or the pulmonary arterial wall and graft interposition.10, 11) In our patient, the RPA had particular features; originating from the left posterior aspect of the proximal AAo and being unusually connected to the PT. Thanks to these, the RPA could be directly reimplanted to the PT without causing stenosis.
In conclusion, we encountered a premature girl, weighing 1,700 g, who had an unusual form of AORPA. The RPA communicated with the PT via a small lumen. Direct reimplantation to the PT was successfully established. She remained asymptomatic with no RPA stenosis in the longer term at five years postoperatively.
Author Contribution
TM contributed to study concept and design and was a major contributor to writing the manuscript. KF, KY and OM contributed to study concept and design, and made contributions to revising the manuscript. KI and HS performed clinical examinations and interpreted the patient data. All authors read and approved the final manuscript.
 1,Keiichi Fujiwara1,Kotaro Inaguma2,Kosuke Yoshizawa1,Otohime Mori1,Hisanori Sakazaki2Toshi Maeda
1,Keiichi Fujiwara1,Kotaro Inaguma2,Kosuke Yoshizawa1,Otohime Mori1,Hisanori Sakazaki2Toshi Maeda