Myocarditis is a disease with many different appearances. It can pass unnoticed or have a mild course or even lead to a serious course with severely impaired cardiac function and heart failure.1) In children, particularly severe courses occur in infants and young children. Adolescents tend to have milder courses of the disease, which has led to the hypothesis of different pathomechanism in the different age groups.2) In addition to age-related differences in immunological response, genetic variants in cardiomyopathy genes are currently being discussed.3, 4) These could be a cause of severe heart failure with impaired function and the phenotype of dilated cardiomyopathy (DCM) in children with myocarditis. However, it is unclear whether these children already had a DCM phenotype prior to the initial presentation and whether an additional infection led to a further deterioration of the condition. Another hypothesis is that the clinical picture of the DCM phenotype only becomes apparent after an infection. The presence of genetic variants in cardiomyopathy genes, however, may then lead to the severe phenotype.5, 6) Little is known about the outcome of children and adolescents with myocarditis in the presence of genetic variants in cardiomyopathy genes. The aim of this review is to provide an overview of the presence of these genetic variants in pediatric myocarditis and their impact on clinical presentation and phenotypes. Whereas myocarditis is defined according to the available histologic, immunologic and immunhistochemic criteria.7) In addition, the impact of the genetic predisposition on the outcome of the disease is highlighted. These findings could improve risk prediction and may influence subsequent therapy management in the context of listing for heart transplantation or weaning from mechanical circulatory support systems. The prospective multicenter registry for children and adolescents with suspected myocarditis “MYKKE” aims to investigate pediatric myocarditis and its course as well as to identify different pathomechanisms. Previously published data on genetic findings of myocarditis patients within the “MYKKE” registry serve as the basis for this review.2, 3, 8)
Heterogeneous Clinical Presentation of Pediatric Myocarditis
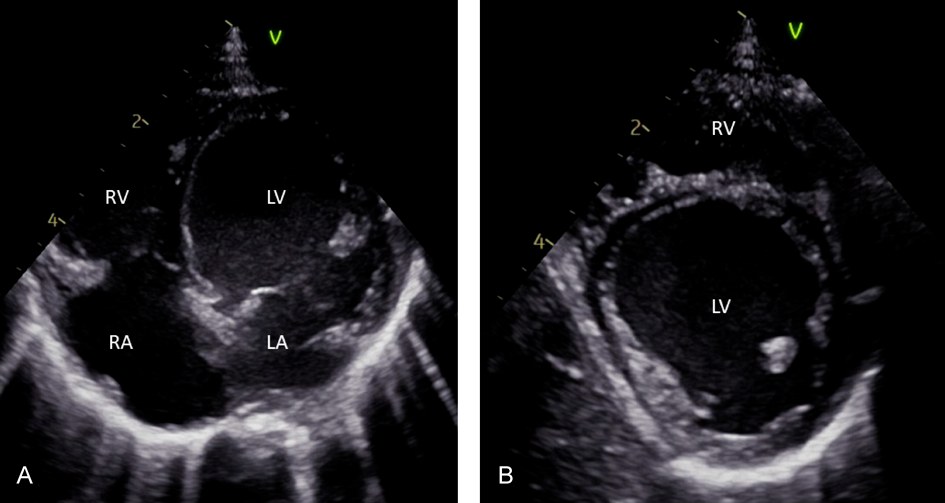
As mentioned above, pediatric myocarditis manifests with varying symptoms and ranges from severe heart failure to a mild disease course with rapid resolution of symptoms. The often non-specific symptoms and their diversity frequently cause a delay in diagnosis.9) The age distribution of pediatric myocarditis reveals two distinct peaks: infancy and adolescence.10, 11) This is accompanied by differences in the clinical presentation. In the multicenter registry “MYKKE”, patients<2 years of age were more likely to have severe left-ventricular impairment and cardiac adverse events like need for mechanical circulatory support and death, with an even sex distribution in this age group (Figs. 1 and 2). This cohort presents with a heart failure-like myocarditis. The probability of severe disease progression declines with age by 14% a year.2, 3, 12) Nearly half of the children with impaired function at initial presentation do not recover fully.13) Another large multicenter study found that moderate to severe ventricular dysfunction was associated with young age, female sex and the presence of respiratory distress and gastrointestinal symptoms (including nausea, emesis, diarrhea, poor appetite).10) The presence of unspecific gastrointestinal symptoms such as emesis and abdominal pain may delay diagnosis of myocarditis in younger children.14) A Japanese survey study found that in pediatric patients with acute and fulminant myocarditis, gastrointestinal symptoms were even more common than cardiorespiratory symptoms at the onset of the disease.15)
In contrast to that, myocarditis in adolescent’s manifests most often with chest pain or acute coronary syndrome-like symptoms. Chest pain was the most common symptom in adolescent patients with preserved or mildly reduced ventricular function.10, 16) Among the adolescents diagnosed with myocarditis, males account for about two-thirds of patients.2) This is consistent with findings of myocarditis in adults, which thus might share similar pathophysiologic mechanisms.17–19)
In the sub-study of the “MYKKE” registry, patients with myocarditis and DCM phenotype with 35% disease-associated genes had a significantly worse event-free survival compared to the group without DCM phenotype, but also compared to a group of children with DCM and no evidence of inflammation. In addition to the genetic background, myocardial inflammation appeared to be an important factor influencing the outcome.3)
Genetic Predisposition in Pediatric Myocarditis
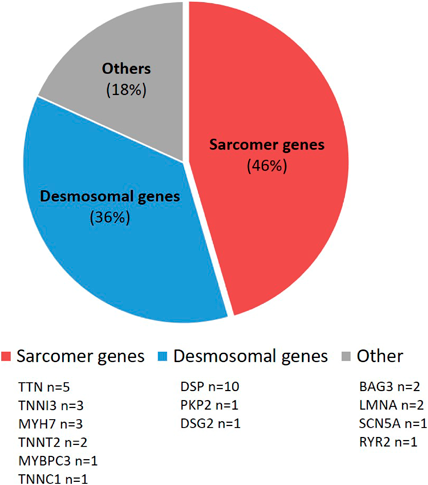
It has been shown that there is an influence of genetic variants in the context of myocarditis.1) The proportion of myocarditis patients with pathogenic variants ranges from 8 to 16% in larger adult cohorts.20, 21) However, the number of pathogenic variants detected increases significantly to 33% with the severity of the myocarditis.22) These are particularly cardiomyopathy-associated genes that affect the sarcomere and desmosome. In addition to the phenotype of DCM, arrhythmogenic right ventricular cardiomyopathy (ARVC) with the occurrence of malignant cardiac arrhythmias also plays a major role in myocarditis, with ARVC being more frequently associated with variants in desmosomal genes.23–25)
The number of cases for genetic variants in children and adolescents with myocarditis is limited (Table 1). Small cohorts show varying frequencies, but again depending on the severity of the disease. Among a cohort of eight children, all of whom were treated for acute heart failure or cardiac arrest at a pediatric intensive care unit, likely pathogenic or pathogenic variants were found in seven of these children. Half of these children were under one year of age. The identified cardiomyopathy-associated variants included titin (TTN, n=2); myosin binding protein C3 (MYBPC3, n=1); Troponin T2 (TNNT2, n=1) and sodium voltage-gated channel alpha subunit 5 (SCN5A, n=1). Two patients had genetically confirmed underlying syndromic diseases (Alström and Hurler syndrome).26) In contrast, in another cohort of seven children with genetic testing and an initial diagnosis of myocarditis, only one single pathogenic variant was found in the Lamin A (LMNA) gene.27) Further, genetic variants in three out of 13 children (23%) with acute myocarditis are reported, with two cases exhibiting multiallelic variants.21)
Table 1 Genetic characterization of pediatric patients with myocarditis| Authors | Year of publication | Patient characteristics (age, sex) | Sample size | Genotype | Cardiac phenotype | Myocarditis type |
|---|
| Brown EE et al.26) | 2019 | 50%<1 y (range 3 weeks to 14 y) 88% female | 8 | TTN n=2;
TNNT2 n=1;
MYBPC3 n=1;
SCN5A n=1;
Syndromic disease n=2 (Alström & Hurler syndrome) | Acute heart failure with reduced LVEF, LV dilatation | Suspected myocarditis at time of presentation |
| Kontorovich AR et al.21) | 2021 | Age<20 y | 13* | Pt 1 (13 y, female): multiallelic TTN
Pt 2 (0.6 y, female): PRDM, DSP, DNM2, DMD (multiallelic)
Pt 3 (19 y, female): DSP | No in depth-characterization | Acute myocarditis with histological evidence |
| Seidel F et al.3) | 2017 | MYC-DCM (n=20): median age 1.4 y; 55% male
MYC-NonDCM (n=22): median age 16 y; 64% male | 42 | 22% with LP/P variants:
MYC-DCM (35%, n=7):
BAG3 n=2
LMNA n=1
MYH7 n=1
TNNI3 n=1
TNNT2 n=1
TTN n=1
MYC-NonDCM (9%, n=2):
DSP n=2 | MYC-DCM: Acute heart failure with reduced LVEF (median 22%), LV dilatation
MYC-NonDCM: chest pain, arrhythmias, preserved LVEF (median 59%) | Myocarditis with histological evidence (acute or chronic/healing) |
| Ammirati E et al.24) | 2022 | Median age 15.5 y; 90% male (n=9) | 10* | DSP n=8
DSG2 n=1
PKP2 n=1 | Chest pain, palpitations, elevated serum troponins, preserved LVEF | Acute myocarditis with evidence in biopsy and/or cardiac MRI |
| van der Meulen MH et al.27) | 2022 | Age<18 y, no further details available | 7* | LMNA n=1 | Acute heart failure with reduced LVEF, LV dilatation | Definite or probable myocarditis |
| BAG3, BAG cochaperone 3; DMD, duchenne muscular dystrophy; DNM2, dynamin 2; DSG2, desmoglein 2; DSP, desmoplakin; LMNA, lamin A; LV, left ventricular; LVEF, LV ejection fraction; MRI, magnetic resonance imaging; MYBPC3, myosin binding protein C3; MYH7, myosin heavy chain 7; PKP2, plakophilin 2; RYR2, ryanodine receptor 2; SCN5A, sodium voltage-gated channel alpha subunit 5; TNNC1, troponin C1; TNNI3, troponin I3; TNNT2, Troponin T2; TTN, titin. * Sample size indicated represents only a sub-cohort in the study, therefore some detailed information on age or sex may not be available. |
In contrast, Ammirati et al. reported 10 children with acute myocarditis and genetic variants in desmosomal genes (desmoplakin (DSP) n=8; plakophilin 2 (PKP2) and desmoglein 2 (DSG2), n=1 respectively), with only one child having a reduced left ventricular ejection fraction (LVEF) of 43%, the others had a preserved LVEF. All had chest pain; four also had palpitations, one a syncope and one presented with additional dyspnea. Cardiac arrhythmia like ventricular tachycardia (VT) was reported in one patient. This group of patients can be clinically classified as having chest pain-like myocarditis, not only due to the described symptoms, but also based on the patients’ ages of 10–17 years. In addition, recurrent myocarditis, a positive family history for myocarditis and distinctive patterns in late enhancement sequences on magnetic resonance imaging with septal enhancement and a ring-like pattern as well as non-sustained VT were found more frequently in the entire presented cohort with pathogenic desmosomal gene variants compared to patients without desmosomal gene variants.24) A sub-study of the “MYKKE” registry examined 42 patients with biopsy-confirmed myocarditis by targeted panel sequencing for cardiomyopathy-associated genes. Likely pathogenic and pathogenic genes were found in nine children (22%). However, they were significantly more frequent in children with the DCM phenotype (MYC-DCM, n=20), who again belonged to the heart failure-like myocarditis group with a median age of 1.4 years. They were compared to a group without DCM phenotype (MYC-NonDCM, n=22) with a median age of 16.1 years. In contrast, the latter group presented with chest pain and ST-segment elevation on electrocardiogram, which once again places them in the chest pain-like myocarditis cohort. Overall, disease-associated variants were identified in seven out of 20 children in the MYC-DCM cohort (35%). These were variants in the genes BAG cochaperone 3 BAG3 (n=2), LMNA, myosin heavy chain 7 (MYH7), troponin I3 (TNNI3), TNNT2 and TTN (n=1, respectively). In two of the patients from the MYC-NonDCM cohort, however, DSP was identified as the disease-associated variant.3)
In a second study, in addition to cardiomyopathy-associated genes, immune disorder genes were examined in 12 children with severe courses of myocarditis and their families. Cardiomyopathy-associated variants were found in the majority (8/12 patients, 67%) of these children. Looking at the immune disorder genes, likely pathogenic variants were also found in three patients (dynein axonemal heavy chain 11 (DNAH11), FA complementation group C (FANCC), and sperm-associated antigen 1 (SPAG1)). Overall, variants in genes associated with ciliary transport were frequently present. The impact of this on the progression of myocarditis is unclear and requires further exploration.8)
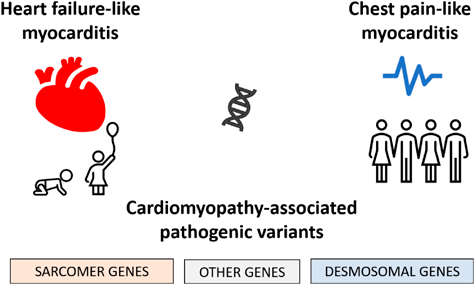
In summary, these results highlight clear clinical and genetic differences in children and adolescents with cardiomyopathy-associated genetic variants and myocarditis (Fig. 3). Overall, infants or young children with heart failure-like myocarditis are more likely to have alterations in sarcomere genes, whereas adolescents with chest pain-like myocarditis and preserved cardiac function are more likely to have genetic variants in desmosomal genes (Fig. 4).
Outcome of Patients with Pediatric Myocarditis and Genetic Background
A remarkable number of pediatric patients with myocarditis requires heart transplantation (17–19%) and the overall mortality in pediatric myocarditis lies around 6%.28) As mentioned earlier, data show that there is an accumulation of genetic variants in severe disease courses.3)
In a pediatric DCM cohort, younger children with likely pathogenic or pathogenic variants were significantly more likely to undergo heart transplantation or decease.27) However, in a mixed cohort of children and adults, no significant difference in clinical outcome was found between patients with and without detected genetic variants. The two children mentioned there with multiallelic genetic cardiomyopathy-associated variants and myocarditis underwent left ventricular assist device implantation and heart transplantation.21)
A similar outcome was found in the eight patients from the Brown et al. cohort. Those with evidence of genetic variants did not show recovery, instead two children died and five other children needed heart transplantation or were listed for transplantation.26)
With regard to desmosomal genes in acute myocarditis and preserved myocardial function of the chest pain-like type myocarditis, it is shown that the incidence of death, heart failure, ventricular tachycardia and myocarditis recurrence is significantly increased in patients with detected variants in desmosomal genes compared to patients without variant detection and to patients without genetic testing at all.24)
Author Contribution
Conceptualization, F.S., H.M. and S.S.; methodology, F.S., D.M., H.M. and S.S.; formal analysis, F.S., and N.R.; investigation, F.S., N.R., T.H., J.H., H.M. and S.S.; resources, F.S., N.R., T.H, J.H., H.M., D.M. and S.S.; writing—original draft preparation, F.S., N.R. and S.S.; writing—review and T.H., J.H., H.M. and D.M.; funding acquisition, F.S., H.M., D.M. and S.S.
 5Franziska Seidel1,2,3,4, Nele Rolfs1,2,4, Tobias Hecht5, Johanna Hummel5, Hendrik Milting6, Daniel Messroghli2,4,7, Stephan Schubert
5Franziska Seidel1,2,3,4, Nele Rolfs1,2,4, Tobias Hecht5, Johanna Hummel5, Hendrik Milting6, Daniel Messroghli2,4,7, Stephan Schubert