Noonan syndrome was first reported by the American pediatric cardiologist Jacqueline Noonan in 1962 and later turned out to actually constitute a group of disorders exhibiting overlapping clinical characteristics. Having been revealed that a series of genetic mutations with similar attributes cause the circumstance, these syndromic disorders are now known collectively as the “RASopathies.” All of the RASopathy-related genes are involved in the regulation of the RAS/MAPK (mitogen-activated protein kinase) signal transduction pathway, and disease-causing mutations lead to hyperactivation of this pathway.1) The RAS/MAPK pathway, as its nomenclature suggests, was initially found as a cardinal signaling cascade for cell proliferation, and is now known to be involved in the multifaceted biological processes of differentiation and cell survival. The spatiotemporal dysregulation of RAS/MAPK activation during embryogenesis can result in a wide range of congenital abnormalities including the cardiovascular system.
The RASopathies are of core importance in the field of pediatric cardiology, comprising over 10% of childhood-onset cardiomyopathy with hypertrophic features, and even up to one-third of the infantile form.2, 3) RASopathy-associated cardiomyopathy (RAS-CMP) differs from primary hypertrophic cardiomyopathy (HCM) that is non-syndromic and isolated; RAS-CMP shows earlier onset and exhibits a more extreme phenotype, such as the propensity towards advanced hypertrophy that affects both left and right ventricles, eventually leading to severe outflow tract obstruction. RAS-CMP is known to be associated with a higher risk of heart failure death. Recent research has shown that RAS-CMP also carries a comparably high risk of sudden arrhythmic death as primary HCM does, while the appropriate preventative measures are less often allocated.4) There is an urgent need for improvement in the quality of care for RASopathies, and this requires a concerted effort from the fields of clinical cardiology and cardiovascular research.
Identifying new causative genes still further in recent years, the RASopathies emerge as an expanding disease entity. This review will focus on the current evidence gaps hampering our molecular understanding of RASopathy pathogenesis, rather than detailing the full clinical spectrum of RASopathies, hoping to set a guide to our next approaches potentially elucidating pathophysiology of the diseases.
Genotype-Phenotype Correlation in RASopathies
RASopathies are, by definition, caused by germline mutations in the molecular components of the RAS/MAPK pathway. Causative genes may encode the RAS molecules, such as HRAS, KRAS, and NRAS, as well as LZTR1,5) a regulator of RAS ubiquitination and quantity control, NF1, a RAS GTPase-activator controlling its cycling between the active and inactive forms, and the downstream effector molecules B/C-RAF, MEK1/2, SHOC2, and SPRED1, among others. The different clinical subtypes of RASopathies include Noonan syndrome, Noonan syndrome with multiple lentigines (NSML), Cardio-facio-cutaneous syndrome, and Costello syndrome. Stemming from the common dysregulation of the RAS/MAPK pathway, it is not surprising to observe shared multi-system abnormalities among different RASopathies, while associated with each RASopathy form are some characteristic features which set the phenotype apart from the others. In this review, we particularly focus on the RASopathy-associated cardiac manifestations, namely RAS-CMP, and discuss how commonly the symptoms can affect RASopathy patients, but with variable frequency and severity. The incidence of RAS-CMP is dependent on the RASopathy subtype, as well as the genotype6); in NSML (caused by mutations in BRAF, RAF1, and PTPN11) and Costello syndrome (caused by mutations in HRAS), RAS-CMP is seen with an incidence as high as 60–80%, while in Noonan syndrome the overall RAS-CMP incidence stays rather low, approximating 20%. Detailing the correlation between the heterogeneous genetic background in Noonan syndrome and the RAS-CMP manifestations, RAF1 and RIT1 mutations are frequently accompanied by RAS-CMP (80 and 50%, respectively), while PTPN11 mutations are less so (10%). When discussing the frequency and severity of RAS-CMP manifestations, it is critical to precisely classify patients and/or diseases not only by the syndromic subtypes, but also by the causative genes (Table 1).7–10)
Table 1 Prevalence of the hypertrophic cardiac phenotype among different RASopathy mutations| Syndrome | Causative gene mutation | Prevalence of cardiac hypertrophy |
|---|
| Noonan syndrome | PTPN11 | 15–20%7, 8) |
| SOS1 | approximately 15%7) |
| RIT1 | approximately 50%7) |
| RAF1 | approximately 80%7) |
| LZTR1 | approximately 50%7) |
| Noonan syndromewith multiple lentigines | PTPN11 | approximately 60%9, 10) |
| BRAF | N.A. |
| RAF1 | N.A. |
| Costello syndrome | HRAS | approximately 80%7) |
| Cardio-facio-cutaneous syndrome | BRAF | 60–100%7, 10) |
| MAP2K1 | N.A. |
| MAP2K2 | N.A. |
Proposed Molecular Mechanisms to Explain the Variability in RASopathy Phenotypic Expression
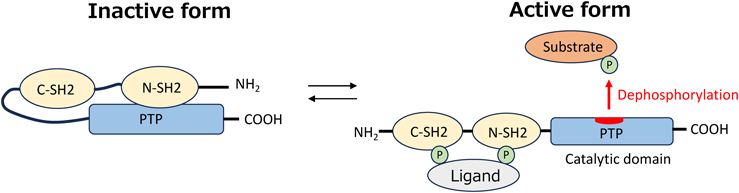
To further consider in depth the variability in RASopathy phenotypes, or in other words the RASopathy genotype-phenotype correlation, we herein take the different PTPN11 mutations for example and discuss their associations with RAS-CMP expression. PTPN11 encodes SHP2, a non-receptor tyrosine phosphatase, which contains two N-terminal Src homology 2 (SH2) domains and a C-terminal protein-tyrosine phosphatase (PTP) domain that exhibits the catalytic function. At steady state, the N-terminal SH2 domains cover the center of catalytic activity, thus maintaining the SHP2 inactive conformation and preventing interaction with its partner molecules and/or substrates. When mitogenic stimuli are transmitted via receptors on the cellular membrane, the SH2 domain binds to the receptor or its scaffold protein, opening up the center of catalytic activity and transforming SHP2 into its active conformation (Fig. 1). PTPN11 is the mutated gene most commonly found in NSML, a RASopathy commonly presenting with severe RAS-CMP, as well as Noonan syndrome, which to the contrary shows lower frequency of RAS-CMP manifestations. Why would the cardiac abnormalities triggered by the same mutated gene show such contrasting phenotypic expression along the RAS-CMP spectrum? The clues to answer this question may be found in the regulatory mechanism of SHP2 phosphatase activity and substrate selectivity. An attractive hypothesis has been presented that the derangements in SHP2 enzymatic activity and the imposed bias in its substrate selection are specific to each PTPN11 mutation, and thus lead to distinct propagating influences upon the downstream signal transduction. The PTPN11 mutations seen in Noonan syndrome are of gain-of-function (GOF) nature. The GOF mutations affect the SH2-PTP interaction, leading to a conformational change favoring exposure of the catalytic center, and result in a constitutively active form of SHP2. The PTPN11 mutations seen in NSML, however, cluster in the catalytic PTP domain destroying its activity and thus are considered loss-of-function (LOF) mutations.11) Changes in SHP2 phosphatase activity may well affect the phosphorylation/dephosphorylation state of its signaling substrate. Bias in substrate selectivity by the mutant SHP2 shall further add complexity to the aberrancy in downstream signaling, namely the RAS/MAPK or the PI3K/AKT pathways. If the RAS-CMP phenotypic expression is to be a summation of the skewed irregularity in the multiple downstream pathways, unveiling the molecular mechanism behind variability in RAS-CMP expression requires a detailed patterning of the downstream deregulation brought about by individual mutations. Intensive efforts are currently in progress to identify SHP2 partner proteins and dephosphorylation targets.12)
Histopathological Hallmarks of RASopathy-Associated Cardiac Hypertrophy: Classic and Current Perspectives
The most recent studies have suggested that RAS-CMPs have poorer cardiac outcomes than primary HCMs. The distinct pathologies behind these two groups of cardiomyopathies shall explain the observed differences in their clinical presentation and prognosis. The classic histopathological hallmark of primary HCMs is myocardial cellular hypertrophy, defined by increase in the cross-sectional area of individual cardiomyocytes. It has long been argued that activation of the RAS/MAPK pathway, acting downstream of the angiotensin and catecholaminergic receptors, contributes to cellular hypertrophy seen in primary HCMs. Since RASopathies are, by definition, a disorder of RAS/MAPK pathway hyperactivation, the natural consequence shall be that the hypertrophied cardiomyocytes comprise the thickened myocardial wall in RAS-CMPs. Indeed, previous histopathological investigations involving RAS-CMPs have documented such cellular hypertrophy of the cardiomyocytes.13, 14) Therefore, substantial overlap in the molecular pathology has been assumed between RAS-CMPs and primary HCMs, and it has generally been held that myocardial cellular hypertrophy is a common trait to both disease entities.
Recently, however, conflicting findings have increasingly been reported. We ourselves have reported on a case with NSML, caused by a LOF mutation in PTPN11, showing fetal-onset myocardial thickening and circulatory failure. At autopsy, it revealed that the thickened myocardium of the patient was not the collective result of individually hypertrophied cardiomyocytes, in other words cellular hypertrophy, but rather associated with an abundancy (∼10% of total) in atypical cardiomyocytes, with reduced cell cross-sectional area, staining positive for the cell proliferation marker Ki67.15) It is known that cardiomyocytes are supposedly terminally-differentiated and have lost their proliferative capacity in exchange of acquiring their unique cellular potential as cardiomyocytes. Therefore, the patient’s autopsy findings reminiscent of cellular ‘hyperplasia,’ at a glance, appeared paradoxical. Following our case, nevertheless, further evidence alluding to the hyperplasia-like phenotype was reported from different animal and cellular models, as well as human cases of RASopathies. It seems that an emerging paradigm for the histopathological hallmarks of RAS-CMPs has begun to take shape. Takahara et al., through their analysis on mice carrying RIT1 (A57G) mutations, have reported myocardial wall thickening, along with an increase in the number of Ki67-positive cardiomyocytes.16) Meier et al. analyzed cardiac histopathology in infants with Noonan syndrome, and their results showed a high proportion of multinucleated cardiomyocytes, indicative of proliferative potential, and also an increase in the number of cardiomyocytes forming the heart. They also analyzed induced pluripotent stem cell-derived cardiomyocytes generated from a RAS-CMP patient harboring a PTPN11 (N308S) mutation, and found similar results indicating an increase in number of multinucleated and Ki67-positive cardiomyocytes.17) Drenckhahn et al. carried out cardiac histopathological analyses on Noonan syndrome patients with RAF1 and PTPN11 mutations, and found an increase in the number of cardiomyocytes forming the thickened myocardial wall.18) The findings collectively suggest that these RAS/MAPK-related mutations harbor the potential of triggering the cardiomyocyte cell-cycle and (re-)stimulating their proliferative potential. Mitosis was thought not to occur in mature adult cardiomyocytes, and to date no similar observations have been reported with primary HCM or secondary cardiomyopathy; hence, it appears that cardiomyocyte hyperplasia is unique to RAS-CMP. Although the hyperplasia-like histopathology has so far only been linked to a subset of mutations, investigating to which extent the aberrant RAS/MAPK signaling in RAS-CMP shall result in excessive cardiomyocyte proliferation appears intriguing.
In addition, understanding the regulatory machinery that converts cardiomyocytes from initiating a hypertrophic response to a hyperplastic response, under aberrant RAS/MAPK signaling, will aid in elucidating its unique pathogenesis. It has been reported that KRAS-null mice show embryonic death due to thinning of the myocardium, and KRAS mutant (V14I) mice exhibit hypertrophic hearts accompanied by cellular hyperplasia. These observations suggest that, whereas HRAS is known for its major involvement in cellular hypertrophy, KRAS may contribute to cardiomyocyte proliferation; however, whether this argument applies to all KRAS mutations needs verification.19, 20) Likewise, a better molecular characterization of these atypically hyperplastic cardiomyocytes that are contributing to the RAS-CMP phenotype, shall map novel drug targets for RAS-CMP. In a study performing single-cell RNA sequencing using cardiac tissue from Noonan syndrome patients, Drenckhahn et al. showed that Noonan syndrome hearts showed an increase in MYH6/MYH7 expression ratio, as well as a reduction in the fatty acid metabolism-related gene expression, both serving as markers of fetal-reprogramming of the cardiomyocyte. A key characteristic of RAS-CMP, therefore, could be the preservation of cardiomyocytes’ characteristics related to immaturity of the cellular phenotype.18) Indeed, immature cardiomyocytes are known to display diminished contractile capabilities and reduced efficiency in energy production and, therefore, may be susceptible to collapsing of compensatory mechanisms when subjected to increased mechanical load. Furthermore, the proposed increase in cellular proliferation and turnover complicating RAS-CMPs could also lead to accumulation of DNA damage through replicative stress, leading to functional deterioration through the so-called ‘premature aging’ of cardiomyocytes. Understanding the effects that aberrant RAS/MAPK signaling has on cardiomyocytes from the perspective of ‘cellular senescence’ is another intriguing approach in light of its downstream histopathological effects including inflammatory or fibrotic changes. Pathological investigations focused on myocardial cellular senescence seem currently underway in an effort to elucidate the unfavorable prognoses observed in RAS-CMP.21) These emerging concepts prompt a recharacterization of the cellular pathologies behind RAS-CMPs.
The Cellular Origin of RAS-CMPs
Since the 1980s, extensive analyses on RAS-CMP mouse models have led to considerable advances in our understanding of the functional role of the RAS/MAPK signaling in cardiac pathology. Especially, the cell type-specific genetically engineered animal models have contributed greatly to pinpointing critical signaling pathways leading to RASopathy-associated cardiac phenotypes, including cardiac hypertrophy, as well as structural heart abnormalities, by identifying the exact cell populations and their intercellular crosstalk that are responsible for the holistic phenotypic expression. Kontaridis et al. reported that mice with cardiomyocyte-specific deletion of PTPN11 show a dilated cardiomyopathy-like phenotype.22) Subsequently, Schramm et al. generated a murine model that selectively expressed mutant PTPN11 in cardiomyocytes, leading to the development of cardiac hypertrophy in these mice.23) These findings suggest that the aberrant signaling taking place endogenously in the cardiomyocytes themselves trigger the onset of RAS-CMP heart lesions, supporting the notion that the cellular origin of RAS-CMP lies within the cardiomyocytes. Conversely, Lauriol et al. found that cardiac hypertrophy was induced in endothelium-specific PTPN11 knock-in mice,24) and they argue that the aberrant RAS/MAPK signaling in the Tie2-positive endothelial cells lead to forming a hypertrophic heart tissue. Araki et al. investigated mice carrying PTPN11 mutations specific to various cell lineages, and found that cardiac septal defects were specifically associated with enhancement in Tie2-positive, endothelial cell ERK signaling and upregulated epithelial-mesenchymal transition.25) Septal defects have also been found in approximately two-thirds of Nkx2.5-positive, cardiac progenitor-specific RAS/MAPK activation.24) In addition, mice with mutant SHP2 expression and downstream ERK activation in the endothelial cell lineages have shown abnormal enhancement in epithelial-mesenchymal transition involving the right ventricular outflow, recapitulating the characteristic right heart defects seen in Noonan syndrome patients.26) A series of studies have not demonstrated thus far that the cellular origin of RAS-CMP is limited to the cardiomyocytes, but have proven suggestive of a diverse multicellular crosstalk, including the endothelium-cardiac progenitor crosstalk, to act upstream during cardiac morphogenesis.
Originating from the first neurofibromatosis type 1 mouse model reported in 1994, a variety of RASopathy animal models using drosophila, mice, and zebrafish have been published. These animal models have clearly shown that intracellular signaling pathways, such as PI3K/AKT/mTOR, as well as the RAS/MAPK pathway, are involved in RAS-CMP pathogenesis. Cardiac hypertrophy associated with RAF1 mutations are induced by downstream ERK activation.27) As for the cardiac hypertrophy in mice harboring a LOF PTPN11 mutation, however, it appears not the ERK signaling but the AKT/mTOR pathway which activates downstream to drive the pathogenesis.23) In addition, in the heart of a zebrafish model of HRAS-mutated Costello syndrome, no activation was seen in either ERK or AKT signaling, but instead findings were suggestive of promoted cellular senescence.28) We may therefore assume that there are numerous pathways other than the RAS/MAPK pathway, the contribution of which are still underestimated, culminating in the full spectrum of RASopathy-associated cardiac pathologies. Each RASopathy-related mutation shall thus trigger the respective pathways to variable extents. A systematic understanding to how the respective downstream signaling act differently in each RASopathy case will be important in assessing pathophysiology, as well as interpreting their responses to pharmaceutical interventions. For example, with the NSML-related PTPN11 LOF mutation that led to over-activation of the mTOR/AKT pathway, the targeted inhibition of mTOR/AKT may offer a greater chance of treatment success than targeting the RAS/MAPK downstream effectors such as MEK—this is in contrast to the NS-related GOF mutation, which mainly activates the RAS/MAPK pathway.
As we have seen, it appears that cell lineages other than cardiomyocytes may influence RAS-CMP pathogenesis. The conundrum therefore remains; what kind of abnormalities in which cell types are actually causing the tissue hypertrophy in RAS-CMPs? A clear conclusion has yet to be reached, but the gaps in knowledge are on the way of becoming fulfilled, as we further stress the importance of subclassifying the RAS-CMP pathologies based on their downstream signaling aberrancies.
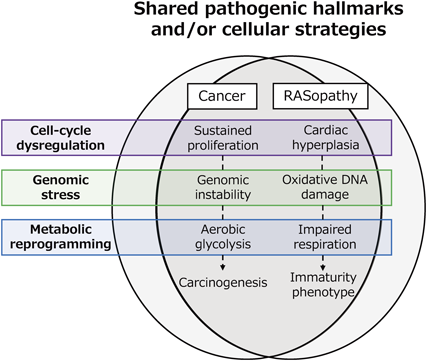
RASopathies as the Intersection of Cancer Biology and Myocardial Pathology
As noted in the introduction of this article, the RAS/MAPK pathway, which was found to be a key signaling pathway in cell proliferation, is involved in multiple aspects of the biological processes of cells, such as differentiation, maturation, senescence, and death. Thus, RAS/MAPK-related genes may serve as the drivers of carcinogenesis through their somatic mutations; mutations in KRAS, followed by NRAS and HRAS, are reported to be involved in various human cancers, while BRAF mutations are found with high frequency in malignant melanomas. In turn, germline mutations of the same genes may affect organogenesis and become the cause of RASopathies. Among the abnormalities common to variable RASopathies are predisposition to specific cancer risks and cardiac disease, including malformations and myocardial disease. RASopathies, residing at the intersection of cancer and cardiac biology, form a unique pathological entity enabling researchers to explore the extent of overlap between cellular pathologies of the heart and cancer tissues. Also, it is of note that when we look at the single nucleotide level, disease-related mutations that lie within the same gene tend to fall into two distinct categories: mutations that contribute strongly to carcinogenesis and mutations that cause RASopathies. According to the hypothesis by a dermatologist Happle, mutations with the most deleterious influence on protein function, so-called “strong alleles,” will not occur as germline mutations, because they are irrevocably detrimental to the survival of the individual if occurring during the embryonic stage. It is thus an intriguing possibility that a similar relationship between allele strength and mutational patterns might exist between cancer- and RASopathy-related mutations.29)
How analogous are the cellular abnormalities in RAS-CMP cardiomyocytes to those in RAS/MAPK-related cancer cells (Fig. 2)? In their review, “The Hallmarks of Cancer,” Weinberg et al. put forward six cardinal traits that define a cancer cell.29) Normal somatic cells acquire these traits sequentially, ultimately leading to complete carcinogenesis. Among these traits, the self-sufficiency in growth signals and insensitivity to antigrowth signals are the important, classic hallmarks of cancer, as cancer cells exhibit autonomous abnormal growth activity that is independent of the environment. As noted above, cardiomyocytic hyperplasia-like changes in the myocardium of different RAS-CMPs have been recently reported. The findings suggest that both the abnormal cancer and cardiac tissues stemming from aberrant RAS/MAPK signaling may share overlapping cellular abnormalities resulting in a similar hyperproliferative phenotype. Cancer cells further harbor instabilities surrounding their genome that promotes the accumulation of somatic mutations, which act to solidify and/or promote the malignant cellular abnormalities including their hyperproliferative traits. In relation to this cancer genome instability, the cardiomyocyte genome similarly exhibits susceptiveness to oxidative damage, being most intensively exposed to oxidative stress in return for the active aerobic respiration fueling the heart. Recent advances in cardiovascular research have shown that, just as genomic instability creates a predisposition to the acquisition of cancer traits, constant exposure of the cardiomyocyte genome to oxidative damage plays a central role in the acquisition of senescent traits in cardiomyocytes and their microenvironment through the accumulation of detrimental mutations.30)
In cancer biology, reprogramming in the energy metabolism is meant to couple the abnormal cellular proliferative activity. In normal somatic cells, glucose-derived pyruvate is either fueled to the mitochondria under aerobic conditions, or converted to lactate under anerobic conditions to balance oxygen consumption. The RAS/MAPK pathway is known as a major factor in the cellular metabolic switching, and the cancer cell’s skewedness towards active use of glycolysis, even under aerobic conditions, is termed aerobic glycolysis. Enhancement of the glycolytic pathway plays a central role in diverting or supplying glycolysis metabolic intermediates to pathways for biosynthesis of nucleotides and amino acids that are essential for the construction of new cells, and this appears to be part of the purposeful survival strategy of cancer cells.31) Such uniqueness in cellular energy metabolism has been best described in Costello syndrome. This form of RASopathy, caused by a heterozygous mutation in HRAS, often presents with severe cardiac hypertrophy. Costello syndrome patients exhibit an exacerbated basal energy expenditure, and a tendency towards hypoglycemia and hypercholesterolemia, accompanied by other derangements in cellular energy metabolism. Costello syndrome model mice with HRAS (G12S) mutation also replicate the metabolic phenotype of human patients, including hypoglycemia and impaired fatty acid oxidation in the mitochondria.32) These findings may be considered as evidence for RAS-CMP metabolic reprogramming that echoes increased glucose utilization seen in RAS/MAPK-related malignancies. The cardiomyocytes’ metabolic phenotype favoring cell proliferation and the cellular hyperplasia observed in RAS-CMPs seems likely to be bridged by a causal relationship. Such reprogramming of energy metabolism has been observed in other RASopathies as well. In NS caused by the PTPN11 mutation, decreased enzymatic activity of the mitochondrial respiratory chain, decrease in cellular ATP content, and increased levels of reactive oxygen species have been reported.33) RASopathies associated with BRAF and NF1 mutations have exhibited impairment in mitochondrial bioenergetics related with reduced mitochondrial respiratory chain activity.34, 35) Whether the causal relationship between RAS/MAPK metabolic reprogramming and RAS-CMP pathology underlies the whole spectrum of RASopathies is yet to be fully clarified.
Understanding the entirety of such RAS-CMP compensatory mechanisms from the viewpoint of its analogies with RAS/MAPK-mediated cancer biology is another attractive approach towards searching for new druggable targets. Internal or external biological stressors such as malignant transformation or invasive pathogens are robustly compensated by the human host through innate cellular defense mechanisms. While the cardiomyocytes in RAS-CMPs shall encounter a spectrum of cellular stressors as a consequence of aberrant RAS/MAPK signaling, our appreciation of the compensatory mechanisms at play remains even more incomplete.
Various molecular-targeted therapies are now increasingly being developed for heart diseases, ushering in a new era of cardiovascular medicine. With RAS-CMP, however, the clinical development of RAS-CMP-specific therapies has currently been outstripped by that for primary HCM. Still, small case series have been reported on the compassionate use of MEK inhibitors (e.g., trametinib) against Noonan syndrome cardiac hypertrophy.33) While it is essential to await results from larger scale clinical trials, momentum is building toward overcoming the unmet need of RAS-CMP-specific molecular therapeutics. As the development of novel anti-cancer drugs that target the RAS/MAPK pathway gain pace, it is highly likely that a portion of these drugs will exert therapeutic potential against RAS-CMPs. The major focus for now includes creating a translational framework for comprehensive clinical data collection and evidence accumulation, from both the bedside and bench, in order to make the leap from elucidating disease pathophysiology to developing novel therapeutics for RAS-CMPs.
謝辞Acknowledgments
This work was supported by a grant from AMED (JP23bm1423007 to Masamichi Ito, JP23wm0325049 to Yu Nakagama and JP22fk0108576 to Yu Nakagama), Miyata Cardiac Research Promotion Foundation (Yu Nakagama), and The Osaka Medical Research Foundation for Interactable Diseases (Yu Nakagama).
Conflicts of Interest
None.
Originally published in Pediatric Cardiology and Cardiac Surgery, Vol. 39 (2023), pp. 192–199. [in japanese] doi: 10.9794/jspccs.39.192
引用文献References
1) Aoki Y, Niihori T, Narumi Y, et al: The RAS/MAPK syndromes: Novel roles of the RAS pathway in human genetic disorders. Hum Mutat 2008; 29: 992–1006
2) Aljeaid D, Sanchez AI, Wakefield E, et al: Prevalence of pathogenic and likely pathogenic variants in the RASopathy genes in patients who have had panel testing for cardiomyopathy. Am J Med Genet A 2019; 179A: 608–614
3) Norrish G, Kolt G, Cervi E, et al: Clinical presentation and long-term outcomes of infantile hypertrophic cardiomyopathy: A European multicentre study. ESC Heart Fail 2021; 8: 5057–5067
4) Lynch A, Tatangelo M, Ahuja S, et al: Risk of sudden death in patients with RASopathy hypertrophic cardiomyopathy. J Am Coll Cardiol 2023; 81: 1035–1045
5) Nakagama Y, Takeda N, Ogawa S, et al: Noonan syndrome-associated biallelic LZTR1 mutations cause cardiac hypertrophy and vascular malformations in zebrafish. Mol Genet Genomic Med 2020; 8: e1107
6) Yaoita M, Niihori T, Mizuno S, et al: Spectrum of mutations and genotype-phenotype analysis in Noonan syndrome patients with RIT1 mutations. Hum Genet 2016; 135: 209–222
7) Leoni C, Blandino R, Delogu AB, et al: Genotype-cardiac phenotype correlations in a large single-center cohort of patients affected by RASopathies: Clinical implications and literature review. Am J Med Genet A 2022; 188: 431–445
8) Ichikawa Y, Kuroda H, Ikegawa T, et al: Cardiac features of Noonan syndrome in Japanese patients. Cardiol Young 2023; 33: 564–569
9) Kim ST, Lee SY, Kim GB, et al: Cardiovascular characteristics and progressions of hypertrophic cardiomyopathy and pulmonary stenosis in RASopathy syndrome in the genomic era. J Pediatr 2023; 262: 113351
10) Kauffman H, Ahrens-Nicklas RC, Calderon-Anyosa RJC, et al: Genotype-phenotype association by echocardiography offers incremental value in patients with Noonan syndrome with multiple lentigines. Pediatr Res 2021; 90: 444–451
11) Kontaridis MI, Swanson KD, David FS, et al: PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J Biol Chem 2006; 281: 6785–6792
12) Zhu P, Wu X, Zhang RY, et al: An integrated proteomic strategy to identify SHP2 substrates. J Proteome Res 2022; 21: 2515–2525
13) Lin AE, Alexander ME, Colan SD, et al: Clinical, pathological, and molecular analyses of cardiovascular abnormalities in Costello syndrome: A Ras/MAPK pathway syndrome. Am J Med Genet A 2011; 155A: 486–507
14) Fahrner JA, Frazier A, Bachir S, et al: A RASopathy phenotype with severe congenital hypertrophic obstructive cardiomyopathy associated with a PTPN11 mutation and a novel variant in SOS1. Am J Med Genet A 2012; 158A: 1414–1421
15) Nakagama Y, Inuzuka R, Ichimura K, et al: Accelerated cardiomyocyte proliferation in the heart of a neonate with LEOPARD syndrome-associated fatal cardiomyopathy. Circ Heart Fail 2018; 11: e004660
16) Takahara S, Inoue SI, Miyagawa-Tomita S, et al: New Noonan syndrome model mice with RIT1 mutation exhibit cardiac hypertrophy and susceptibility to β-adrenergic stimulation-induced cardiac fibrosis. EBioMedicine 2019; 42: 43–53
17) Meier AB, Raj Murthi S, Rawat H, et al: Cell cycle defects underlie childhood-onset cardiomyopathy associated with Noonan syndrome. iScience 2021; 25: 103596
18) Drenckhahn JD, Nicin L, Akhouaji S, et al: Cardiomyocyte hyperplasia and immaturity but not hypertrophy are characteristic features of patients with RASopathies. J Mol Cell Cardiol 2023; 178: 22–35
19) Hernández-Porras I, Fabbiano S, Schuhmacher AJ, et al: K-RasV14I recapitulates Noonan syndrome in mice. Proc Natl Acad Sci USA 2014; 111: 16395–16400
20) Wong JC, Perez-Mancera PA, Huang TQ, et al: KrasP34R and KrasT58I mutations induce distinct RASopathy phenotypes in mice. JCI Insight 2020; 5: e140495
21) Engler M, Fidan M, Nandi S, et al: Senescence in RASopathies, a possible novel contributor to a complex pathophenoype. Mech Ageing Dev 2021; 194: 111411
22) Kontaridis MI, Yang W, Bence KK, et al: Deletion of Ptpn11 (Shp2) in cardiomyocytes causes dilated cardiomyopathy via effects on the extracellular signal-regulated kinase/mitogen-activated protein kinase and RhoA signaling pathways. Circulation 2008; 117: 1423–1435
23) Schramm C, Fine DM, Edwards MA, et al: The PTPN11 loss-of-function mutation Q510E-Shp2 causes hypertrophic cardiomyopathy by dysregulating mTOR signaling. Am J Physiol Heart Circ Physiol 2012; 302: H231–H243
24) Lauriol J, Cabrera JR, Roy A, et al: Developmental SHP2 dysfunction underlies cardiac hypertrophy in Noonan syndrome with multiple lentigines. J Clin Invest 2016; 126: 2989–3005
25) Araki T, Chan G, Newbigging S, et al: Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc Natl Acad Sci USA 2009; 106: 4736–4741
26) Krenz M, Gulick J, Osinska HE, et al: Role of ERK1/2 signaling in congenital valve malformations in Noonan syndrome. Proc Natl Acad Sci USA 2008; 105: 18930–18935
27) Wu X, Simpson J, Hong JH, et al: MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J Clin Invest 2011; 121: 1009–1025
28) Santoriello C, Deflorian G, Pezzimenti F, et al: Expression of H-RASV12 in a zebrafish model of Costello syndrome causes cellular senescence in adult proliferating cells. Dis Model Mech 2009; 2: 56–67
29) Castel P, Rauen KA, McCormick F: The duality of human oncoproteins: drivers of cancer and congenital disorders. Nat Rev Cancer 2020; 20: 383–397
30) Ko T, Fujita K, Nomura S, et al: Quantification of DNA damage in heart tissue as a novel prediction tool for therapeutic prognosis of patients with dilated cardiomyopathy. JACC Basic Transl Sci 2019; 4: 670–680
31) Warburg O: On the origin of cancer cells. Science 1956; 123: 309–314
32) Dard L, Hubert C, Esteves P, et al: HRAS germline mutations impair LKB1/AMPK signaling and mitochondrial homeostasis in Costello syndrome models. J Clin Invest 2022; 132: e131053
33) Lee I, Pecinova A, Pecina P, et al: A suggested role for mitochondria in Noonan syndrome. Biochim Biophys Acta Mol Basis Dis 2010; 1802: 275–283
34) Kleefstra T, Wortmann SB, Rodenburg RJ, et al: Mitochondrial dysfunction and organic aciduria in five patients carrying mutations in the Ras-MAPK pathway. Eur J Hum Genet 2011; 19: 138–144
35) Masgras I, Ciscato F, Brunati AM, et al: Absence of neurofibromin induces an oncogenic metabolic switch via mitochondrial ERK-mediated phosphorylation of the chaperone TRAP1. Cell Rep 2017; 18: 659–672
 2,3,Yu Nakagama
2,3,Yu Nakagama