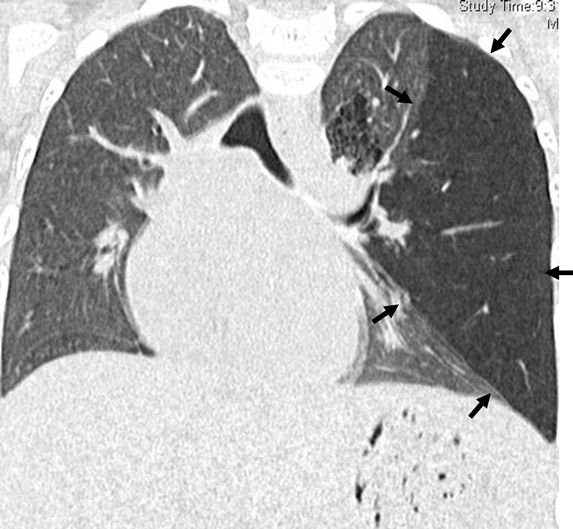
A Pediatric Case of Pulmonary Hypertension Associated with Congenital Bronchial Atresia
1 Division of Cardiology, National Center for Child Health and Development ◇ Tokyo, Japan
2 Division of Respiratory Medicine, National Center for Child Health and Development ◇ Tokyo, Japan
受付日:2024年10月29日Received: October 29, 2024
受理日:2025年4月18日Accepted: April 18, 2025
発行日:2025年7月31日Published: July 31, 2025